
Superhard-driven search of the covalent network in the B3NO system
Abstract
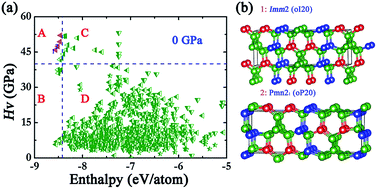
The search for new superhard materials with a Vickers hardness larger than 40 GPa remains a considerable experimental and theoretical challenge. Here, we perform a superhard-driven search using an unbiased structure search method based on the CALYPSO method in the ternary B–N–O system, B3NO, which is isoelectronic with diamond. A variety of newly predicted structures of the B3NO compound with short, strong, and three-dimensional covalent bonds were designed. Among them, two newly predicted orthorhombic structures with Imm2 (oI20) and Pmn21 (oP20) space groups were found to be superhard and energetically stable. After examining the dynamical stabilities, we found that these two structures are energetically more preferable. Further hardness calculations showed that the two structures are superhard materials with a Vickers hardness above 45 GPa, exceeding the criterion of superhard materials. The electronic results show that the oI20 and oP20 structures are semiconductor materials with an optimal band gap of 0.87 and 0.12 eV, respectively. The present results reveal that the B3NO compounds can be used as superhard materials or narrow band-gap semiconductor materials, and therefore have broad prospects in industrial applications, and also provide insights for exploring other functional compounds with a functionality-driven design.
 Please wait while we load your content...
Please wait while we load your content...

