
Ultrasonic enhanced synthesis of multi-walled carbon nanotube supported Pt–Co bimetallic nanoparticles as catalysts for the oxygen reduction reaction
Shaofang Fua,
Guohai Yanga,
Yazhou Zhoua,
Horng-Bin Panb,
Chien M. Waib,
Dan Dua and
Yuehe Lin*ac
aDepartment of Mechanical and Material Engineering, Washington State University, Pullman, WA 99163, USA
bDepartment of Chemistry, University of Idaho, Moscow, ID 83844, USA
cPacific Northwest National Laboratory, Richland, Washington 99352, USA. E-mail: yuehe.lin@wsu.edu
First published on 31st March 2015
Abstract
Carbon material supported bi- or tri-metallic nanoparticles were usually used to replace noble metals, such as platinum, for improving catalytic performance and reducing the cost. In this paper, a carboxylate-functionalized multi-walled carbon nanotube supported bimetallic platinum–cobalt nanoparticles catalyst was synthesized using a simple one-step ultrasonic method. Electrochemical experiments showed that this catalyst exhibited excellent electrocatalytic activity in acid solution for the oxygen reduction reaction. In detail, the onset potential and half-wave potential of this catalyst positively shifted compared with the commercial platinum/carbon catalyst. The as-prepared catalyst also presented a high mass activity. Additionally, it showed a four-electron reduction pathway for the oxygen reduction reaction and exhibited better stability (about 82.8% current density was maintained) than platinum/carbon during the current durability test.
Introduction
Polymer electrolyte fuel cells (PEMFCs) are a leading candidate to replace the aging alkaline fuel cells due to their compact construction, high current density, low operation temperature, and fast start-up.1 To some extent, PEMFCs are considered as efficient and clean energy sources for portable and stationary applications.2,3 However, there are still some challenges which need to be addressed. One of the major challenges impeding the further development of the industrial applications is improving the activity of cathodic catalysts toward the oxygen reduction reaction (ORR).4–8 Carbon material supported platinum nanoparticles (Pt NPs) have been traditionally used in catalysts for the sluggish ORR in PEMFCs because of the high activity of Pt toward ORR. However, Pt NPs tend to dissolve and agglomerate under severe cathode conditions. Besides, the high cost of Pt limits the wide scale usage of PEMFCs as alternative energy sources.9–11To solve these problems, extensive efforts have been made to develop the low-Pt electrocatalysts for fuel cells. Many studies have demonstrated that Pt-based bi- or tri-metallic alloyed electrocatalysts, such as alloying Pt with transition metals, such as iron (Fe),12–14 cobalt (Co),15–20 nickel (Ni),12,14,16,18 chrome (Cr),18,20 vanadium (V),12,20 could improve the durability and activity for the ORR by decreasing the activation energy. Consequently, the amount of Pt is also reduced significantly. On the other hand, a lot of researches have been focused on the support materials to further increase the electrocatalytic property and efficiency. Carbon nanotubes (CNTs) attract much interest as catalyst support due to their high electrical conductivity, high surface area and good durability.21,22 However, the pristine CNTs contain only few binding sites to anchor metal NPs because of their high curvature and inertness. In order to have the good attachment and dispersion of metal NPs, CNTs are usually treated with covalent or non-covalent functionalization.23 Our group has been working on experimental and theoretical studies of CNTs supported metal nanoparticles for several years.23–25 The as-synthesized catalysts exhibit excellent catalytic activities on PEMFCs.
Ultrasonic method has been applied in organic synthesis, materials chemistry, and industrial processes due to its extreme transient conditions, which are distinct with traditional synthesis methods.26–30 It was demonstrated to be a facile method to synthesize nanostructured metal particles. These unique conditions of ultrasound allow the synthesis of materials in a liquid with room temperature, which otherwise requires high temperature, high pressure or long reaction time.26
Herein, carboxylate-functionalized multi-walled CNTs (MWCNTs) supported bimetallic Pt–Co NPs (PtCo/MWCNTs) catalyst was synthesized using a simple one-step ultrasonic method. The electrochemical studies in acid solution showed that PtCo/MWCNTs presented better electrocatalytic activity for ORR through a four-electron reduction pathway compared with commercial platinum/carbon (Pt/C) catalyst. For the stability, 82.8% initial current density was maintained during the test, which is also higher than that of commercial Pt/C catalyst.
Results and discussion
Characterization of PtCo/MWCNTs
The morphology of the product was obtained by transmission electron microscopy (TEM) as shown in Fig. 1a. It reveals that the Pt–Co NPs were uniformly deposited onto the surface of MWCNTs. The formation of Pt–Co bimetallic NPs on MWCNTs support was attributed to the functional groups of the MWCNTs, which served as the anchor sites and bound with NPs by chemical bonds.31 The particle size distribution was calculated by Nano Measure software, which is shown in Fig. 1b. It reveals that the particles are distributed uniformly with an average size around 1.6 nm. | ||
| Fig. 1 (a) TEM image of the PtCo/MWCNTs. (b) Pt–Co NPs size distribution. | ||
The crystalline and lattice properties of the PtCo/MWCNTs catalyst were characterized by X-ray diffraction (XRD), as shown in Fig. 2. The first diffraction peak (2θ = 26°) indicates the graphitic carbon of MWCNTs. A well-defined (111) plane can also be observed at 2θ = 40.33°. These peaks slightly shifted to higher angles than those for Pt compared with the database for pure Pt and Co (e.g., 2θ values of 39.1 (111) for Pt; 44.2 (111) for Co), which demonstrated the alloy formation among Pt and Co. The PtCo NPs size can be calculated according to Scherrer equation:32

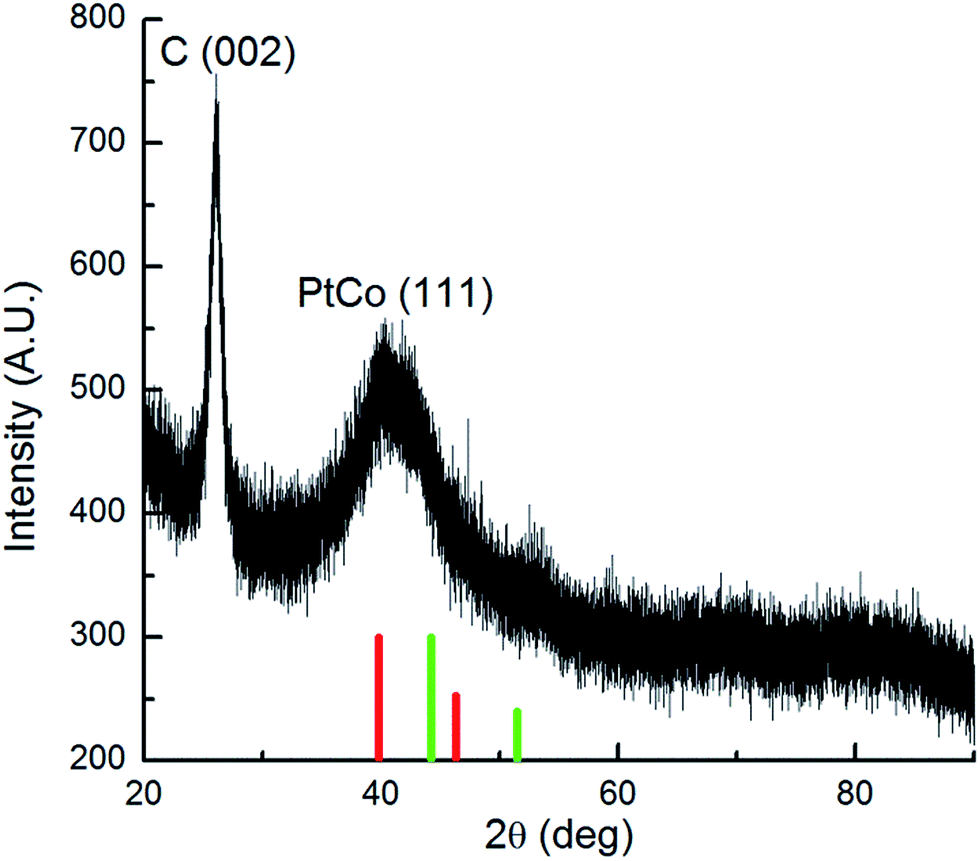 | ||
| Fig. 2 X-ray diffraction pattern of PtCo/MWCNTs. JADE database peak positions for Pt (red) and Co (green) are marked. | ||
The surface composition and interaction between Pt and Co atoms in the PtCo/MWCNTs were studied by X-ray photoelectron spectroscopy (XPS). Fig. 3a and b show the XPS spectra of the sample in Pt 4f and Co 2p regions. The peaks at binding energies of 71.64 eV, 74.79 eV and 780.67 eV correspond to Pt (4f7/2), Pt (4f5/2) and Co (2p3/2), respectively. It reveals that Pt 4f peaks shift to higher binding energy compared to pure Pt NPs,33 indicating electron transfer from Pt to Co. The electronic structures of PtCo NPs are modified due to the alloying of Pt with Co.10,34,35
 | ||
| Fig. 3 XPS spectra of Pt 4f (a) and Co 2p (b) of PtCo/MWCNTs. | ||
Electrocatalytic activity of PtCo/MWCNTs catalysts
In order to evaluate the specific electrocatalytic performance of PtCo/MWCNTs towards ORR, cyclic voltammetry (CV) tests were carried out in N2 and O2-saturated 0.5 M HClO4 solution from 0 V to 1.0 V (vs. Ag/AgCl) at a scan rate of 50 mV s−1. As shown in Fig. 4a, featureless voltammetric currents within the potential range were observed for all electrodes in N2-saturated solution. In contrast, when the solution was saturated with O2, all the CV curves presented well-defined characteristic ORR peaks, indicating the electrocatalytic activities of PtCo/MWCNTs towards ORR. In detail, the peak potential of PtCo/MWCNTs positively shifted (around 40 mV) relative to commercial Pt/C catalyst. The linear sweep voltammetry (LSV) curves for the ORR obtained with PtCo/MWCNTs and Pt/C electrocatalysts on a rotating disk electrode (RDE) in O2-saturated 0.5 M HClO4 solution at a rotating rate of 1600 rpm are shown in Fig. 4b. The onset potential of PtCo/MWCNTs is nearly 40 mV higher than that of commercial Pt/C. A half-wave potential, E1/2, is often used to evaluate the electrocatalytic activity of a catalyst. As shown in Fig. 4b, the half-wave potential of PtCo/MWCNTs (0.763 V) is positively shifted relative to Pt/C catalyst (0.73 V). These results are comparable or even better than that in some reports.6,10 The ORR mass activity of PtCo/MWCNTs catalyst is higher than that of commercial Pt/C as shown in Fig. 4c. All these results indicate the good electrocatalytic activity of PtCo/MWCNTs towards ORR. This might be attributed to the formation of Pt–Co alloy which increases the ORR activity because of the different band configuration between Pt–Co alloy and pure Pt.36,37 In addition, the addition of Co also has an effect on the catalyst, such as geometric structure, particle size, wettability, and so on.4,15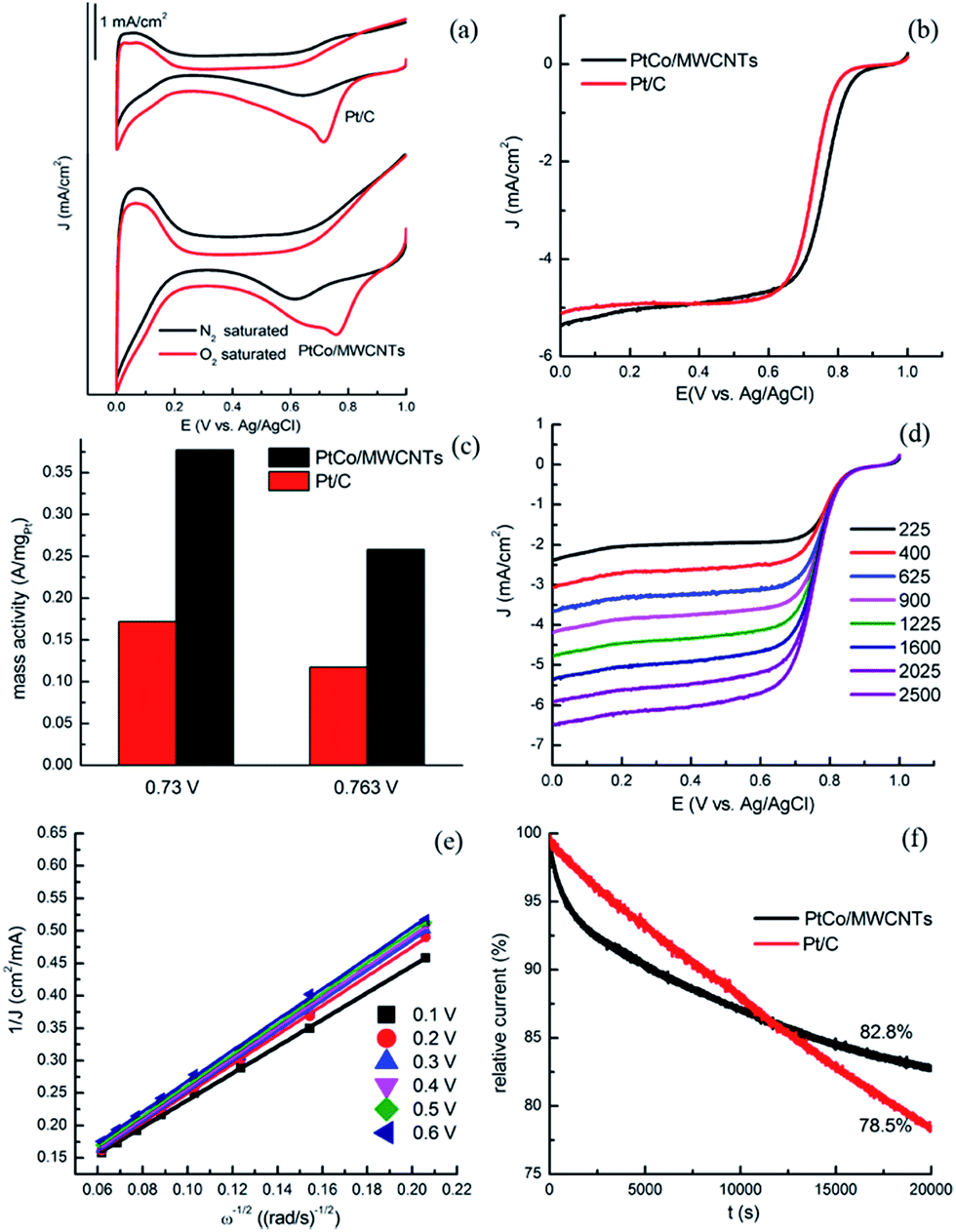 | ||
| Fig. 4 CV curves of PtCo/MWCNTs and commercial Pt/C on RDE in N2-saturated and O2-saturated 0.5 M HClO4 solution at a scan rate of 50 mV s−1. (b) LSV curves of PtCo/MWCNTs and Pt/C electrodes in O2-saturated 0.5 M HClO4 solution at a scan rate of 10 mV s−1 and a rotation rate of 1600 rpm. (c) ORR mass activity of PtCo/MWCNTs and commercial Pt/C catalysts at 0.73 V and 0.763 V vs. Ag/AgCl. (d) LSV curves with various rotation rates. (e) Koutecky–Levich plots of PtCo/MWCNTs electrodes at different potential. (f) Chronoamperometric responses of PtCo/MWCNTs and commercial Pt/C electrodes at 0.3 V vs. Ag/AgCl in O2-saturated 0.5 M HClO4 solution at a rotation rate of 200 rpm. | ||
Further insight of the electron transfer kinetics of ORR process on these catalysts was studied by LSV on an RDE. The rotating speeds were from 225 rpm to 2500 rpm with a scan rate of 10 mV s−1 in O2-saturated 0.5 M HClO4 electrolyte. As shown in Fig. 4d, the current densities increased with rotating rate significantly for PtCo/MWCNTs electrode due to the faster oxygen flux to electrode surface under high rotating rate. ORR precedes either via a two-electron reduction pathway in which hydrogen peroxide is formed as an intermediate or via a four-electron reduction pathway where O2 is directly reduced to OH−. The four-electron pathway is generally preferred in fuel cell since it gives a faster reduction rate. Fig. 4e shows Koutecky–Levich plots of PtCo/MWCNTs at different potentials. Each plot shows a good linear relationship between J−1 and ω−1/2, revealing the consistent electron-transfer at different potential and the first-order reaction kinetics with respect to the concentration of dissolved O2. According to the slopes of the Koutecky–Levich plots, the number of electrons transferred for ORR on PtCo/MWCNTs electrode was calculated to be around four, indicating the nearly complete reduction of O2 to H2O. To demonstrate the stability of PtCo/MWCNTs catalyst, a comparison test with commercial Pt/C was performed by chronoamperometry at a constant voltage of 0.3 V vs. Ag/AgCl in O2-saturated 0.5 M HClO4 electrolyte at a rotating rate of 200 rpm. As shown in Fig. 4f, about 82.8% relative current was persisted after reaction for 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s. In contrast, only about 78.5% of the initial current density was maintained for commercial Pt/C. This confirmed that PtCo/MWCNTs catalysts exhibit a much better stability than commercial Pt/C catalyst for ORR in acid media due to the higher stability of Pt–Co alloy compared to Pt.
000 s. In contrast, only about 78.5% of the initial current density was maintained for commercial Pt/C. This confirmed that PtCo/MWCNTs catalysts exhibit a much better stability than commercial Pt/C catalyst for ORR in acid media due to the higher stability of Pt–Co alloy compared to Pt.
Experimental
Chemicals and reagents
Commercial platinum/carbon (Pt/C) 20 wt% (Pt loading: 20 wt%, Pt on carbon black) and sodium borohydride (NaBH4) were purchased from Alfa Aesar. Potassium chloride (KCl, 99.0–100.5%), 2-propanol (C3H8O, 99.5%), nafion perfluorinated resin solution (5 wt% in mixture of lower aliphatic alcohols and water, contains 45% water), potassium hexachloroplatinate(ii) (K2PtCl4), and perchloric acid (HClO4) were obtained from Sigma-Aldrich, Co. Ethanol (C2H6O, 200 proof) was purchased from Decon Laboratories, INC. and cobalt(II) chloride hexahydrate (CoCl2·6H2O) was purchased from Fisher Scientific. Carboxylic acid functionalized multiwall carbon nanotubes (C-MWCNTs, 10–30 nm in diameter) with purity of 95% were obtained from Nanostructured & Amorphous Materials Inc, USA.Characterization
Transmission electron microscopy (TEM) images were obtained by Philips CM200 UT (Field Emission Instruments, USA). X-ray diffraction (XRD) analysis was operated by Siemens D500, equipped a Cu Kα X-ray source. The tube was operated at 35 kV accelerating voltage and 30 mA current. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos AXIS-165 multi-technique electron spectrometer system with a base pressure of 1 × 10−9 torr. The spectra of the surfaces were obtained with an AXIS-165 manufactured by Kratos Analytical Inc. (Spring Valley, NY, USA) using a monochromatic X-ray radiation of 1487 eV (AlKα). The spectrometer was calibrated against both the Au 4f7/2 peak at 84.0 eV and the Ag 3d5/2 peak at 368.3 eV. Static charging when present was corrected with a neutralizer (flood gun) by placing the carbon peak (C 1s) at about 285 eV.Ultrasonic synthesis of PtCo–MWCNTs
The PtCo/MWCNTs catalyst was synthesized by a simple ultrasonic method. Briefly, 20 mg carboxylic acid functionalized MWCNTs were added into 20 mL ethanol and then the solution was sonicated using a mechanical ultrasonic cleaner bath FS60H (Fisher Scientific, USA) for 1 h to disperse the MWCNTs. After that, 400 μL of a solution containing 200 μL 0.1 M K2PtCl4 and 200 μL 0.1 M CoCl2·6H2O (1:1 molar ratio of Pt2+ and Co2+ ions) were mixed and added into the well-dispersed MWCNTs solution. Then, 15 mg of NaBH4 was added into this mixed solution, followed by 20 min sonication. Finally, PtCo/MWCNTs were obtained by centrifugation and drying at 80 °C for 12 hours. Inductively coupled plasma (ICP) results showed that the molar ratio of Pt and Co in PtCo/MWCNTs system is 1:0.92, and the mass fractions of Pt and Co were 9.1% and 2.5%, respectively. The total metal weight percentage was 11.6%.
Electrode preparation and electrochemical measurements
The Pt/C catalyst (2 mg mL−1) was prepared by dissolving commercial Pt/C into aqueous solution which contains 2 mL 2-proponal, 8 mL deionized water and 0.05 mL nafion. The mixture was ultrasonicated to obtain homogeneous catalyst ink.The PtCo/MWCNTs catalyst (2 mg mL−1) was prepared by PtCo/MWCNTs with 0.25% nafion which was diluted by ethanol. The mixture was ultrasonicated to obtain homogeneous catalyst ink.
The electrochemical measurements were conducted on an electrochemical workstation (CHI 630E) coupled with a three-electrode system. A Pt wire and a Ag/AgCl electrode filled with 3 M KCl aqueous solution were used as the counter electrode and reference electrode, respectively. To prepare the working electrode for ORR and methanol crossover test, 10 μL PtCo/MWCNTs catalyst ink was loaded on a glassy carbon (GC) disk electrode (5 mm in diameter, 0.19625 cm2 in geometric area) and dried at 140 F in vacuum. Pt/C on GC electrodes was prepared in the same way. And the calculated loading was 101.9 μg cm−2. The electrolyte was 150 mL 0.5 M HClO4 solution for ORR test.
Measurements on rotating dick electrode (RDE) were carried out on an electrode rotator (Princeton Applied Research). Cyclic voltammetry (CV) measurements with scan rate of 50 mV s−1 for both ORR and methanol crossover tests were performed after purging nitrogen or oxygen into HClO4 solution for 30 min. Linear sweep voltammetry (LSV) measurements were performed in O2-saturated electrolyte for ORR test. The scan rate was 10 mV s−1.
Conclusions
In summary, MWCNTs supported bimetallic Pt–Co alloy NPs were synthesized by one-step ultrasonic method. The Pt–Co NPs were uniformly distributed on the surface of MWCNTs. The electrochemical experiments showed that PtCo/MWCNTs catalysts presented higher activity towards ORR than that of commercial Pt/C. It also showed a four-electron reduction pathway. Additionally, the PtCo/MWCNTs catalysts showed better stability in the current durability test compared to commercial Pt/C.Acknowledgements
This work was supported by a startup fund of Washington State University, USA. We thank Franceschi Microscopy & Image Center at Washington State University for TEM measurements. Pacific Northwest National Laboratory is a multi-program national laboratory operated for DOE by Battelle under Contract DE-AC05-76RL01830.Notes and references
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2, 864–890 CrossRef CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- M. S. Saha, Y. Chen, R. Li and X. Sun, Asia-Pac. J. Chem. Eng., 2009, 4, 12–16 CrossRef CAS.
- R. Callejas-Tovar and P. B. Balbuena, J. Phys. Chem. C, 2012, 116, 14414–14422 CAS.
- S. Y. Huang, P. Ganesan and B. N. Popov, ACS Catal., 2012, 2, 825–831 CrossRef CAS.
- Z. J. Zhu, Y. L. Zhai and S. J. Dong, ACS Appl. Mater. Interfaces, 2014, 6, 16721–16726 CAS.
- J. A. Wittkopf, J. Zheng and Y. S. Yan, ACS Catal., 2014, 4, 3145–3151 CrossRef CAS.
- S. I. Choi, M. H. Shao, N. Lu, A. Ruditskiy, H. C. Peng, J. Park, S. Guerrero, J. G. Wang, M. J. Kim and Y. N. Xia, ACS Nano, 2014, 8, 10363–10371 CrossRef CAS PubMed.
- S. Takenaka, H. Miyamoto, Y. Utsunomiya, H. Matsune and M. Kishida, J. Phys. Chem. C, 2014, 118, 774–783 CAS.
- Y. G. Zhao, J. J. Liu, Y. H. Zhao and F. Wang, Phys. Chem. Chem. Phys., 2014, 16, 19298–19306 RSC.
- C. H. Cui, L. Gan, H. H. Li, S. H. Yu, M. Heggen and P. Strasser, Nano Lett., 2012, 12, 5885–5889 CrossRef CAS PubMed.
- H. Yano, M. Kataoka, H. Yamashita, H. Uchida and M. Watanabe, Langmuir, 2007, 23, 6438–6445 CrossRef CAS PubMed.
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, J. Power Sources, 2006, 160, 957–968 CrossRef CAS PubMed.
- H. Y. Zhu, S. Zhang, S. J. Guo, D. Su and S. H. Sun, J. Am. Chem. Soc., 2013, 135, 7130–7133 CrossRef CAS PubMed.
- G. Sievers, S. Mueller, A. Quade, F. Steffen, S. Jakubith, A. Kruth and V. Brueser, J. Power Sources, 2014, 268, 255–260 CrossRef CAS PubMed.
- R. Loukrakpam, J. Luo, T. He, Y. S. Chen, Z. C. Xu, P. N. Njoki, B. N. Wanjala, B. Fang, D. Mott, J. Yin, J. Klar, B. Powell and C. J. Zhong, J. Phys. Chem. C, 2011, 115, 1682–1694 CAS.
- Y. Yamada, K. Miyamoto, T. Hayashi, Y. Iijima, N. Todoroki and T. Wadayama, Surf. Sci., 2013, 607, 54–60 CrossRef CAS PubMed.
- M. K. Min, J. H. Cho, K. W. Cho and H. Kim, Electrochim. Acta, 2000, 45, 4211–4217 CrossRef CAS.
- C. S. Rao, D. M. Singh, R. Sekhar and J. Rangarajan, Int. J. Hydrogen Energy, 2011, 36, 14805–14814 CrossRef CAS PubMed.
- D. L. Wang, H. L. L. Xin, R. Hovden, H. S. Wang, Y. C. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- E. Lam and J. H. T. Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef CAS.
- S. Zhang, Y. Y. Shao, G. P. Yin and Y. H. Lin, J. Mater. Chem. A, 2013, 1, 4631–4641 CAS.
- S. Zhang, Y. Y. Shao, G. P. Yin and Y. H. Lin, J. Mater. Chem., 2010, 20, 2826–2830 RSC.
- Y. Y. Shao, R. Kou, J. Wang, C. M. Wang, V. Viswanathan, J. Liu, Y. Wang and Y. H. Lin, J. Nanosci. Nanotechnol., 2009, 9, 5811–5815 CrossRef CAS PubMed.
- Y. H. Lin, X. L. Cui, C. H. Yen and C. M. Wai, Langmuir, 2005, 21, 11474–11479 CrossRef CAS PubMed.
- H. X. Xu, B. W. Zeiger and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 2555–2567 RSC.
- Z. J. Zhang and J. P. Li, Rare Met. Mater. Eng., 2012, 41, 1700–1705 CrossRef CAS.
- C. T. Hsieh and J. Y. Lin, J. Power Sources, 2009, 188, 347–352 CrossRef CAS PubMed.
- R. Ahmadi, M. K. Amini and J. C. Bennett, J. Catal., 2012, 292, 81–89 CrossRef CAS PubMed.
- J. Qi, L. Xin, Z. Y. Zhang, K. Sun, H. Y. He, F. Wang, D. Chadderdon, Y. Qiu, C. H. Liang and W. Z. Li, Green Chem., 2013, 15, 1133–1137 RSC.
- H. B. Pan and C. M. Wai, J. Phys. Chem. C, 2009, 113, 19782–19788 CAS.
- H. B. Pan and C. M. Wai, New J. Chem., 2011, 35, 1649–1660 RSC.
- J. F. Xu, X. Y. Liu, Y. Chen, Y. M. Zhou, T. H. Lu and Y. W. Tang, J. Mater. Chem., 2012, 22, 23659–23667 RSC.
- J. G. Zhang, H. Asakura, J. van Rijn, J. Yang, P. Duchesne, B. Zhang, X. Chen, P. Zhang, M. Saeys and N. Yan, Green Chem., 2014, 16, 2432–2437 RSC.
- J. G. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- H. L. Peng, F. Liu, X. Liu, S. Liao, C. You, X. Tian, H. Nan, F. Luo, H. Song, Z. Fu and P. Huang, ACS Catal., 2014, 4, 3797–3805 CrossRef CAS.
- H. L. Peng, Z. Y. Mo, S. J. Liao, H. G. Liang, L. J. Yang, F. Luo, H. Y. Song, Y. L. Zhong and B. Q. Zhang, Sci. Rep., 2013, 3, 1765 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |