
A solvent evaporation route towards fabrication of hierarchically porous ZSM-11 with highly accessible mesopores
Wen Songa,
Zhiting Liua,
Liping Liub,
Anne Ladegaard Skovc,
Nan Songa,
Guang Xiong*b,
Kake Zhu*a and
Xinggui Zhoua
aUNILAB, State Key Lab of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, 130 Meilong road, Shanghai 200237, P. R. China. E-mail: kakezhu@ecust.edu.cn
bState Key Lab of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, 2 Lingshui road, Dalian, Liaoning 116024, P. R. China. E-mail: gxiong@dlut.edu.cn
cCenter for Energy Resources Engineering, Department of Chemical and Biochemical Engineering, Technical University of Denmark, Søltofts Plads, DK-2800, Kgs. Lyngby, Denmark
First published on 26th March 2015
Abstract
A route to generate hierarchically porous zeolite ZSM-11 has been paved via solvent evaporation induced self-assembly assisted by hexadecyltrimethoxysilane to produce a preformed dry gel, followed by its subsequent transformation into zeolite via steam-assisted-crystallization. The crystallization in dry gel has been found to undergo an orientated attachment growth mechanism whereby hexadecyltrimethoxysilane directs the formation of auxiliary mesopores and inhibits the fusion of primary nucleates. Measurements such as XRD, SEM, TEM, N2-physisorption, and TEM for an inverse replica of Pt derived from hierarchical ZSM-11 have been conducted to characterize the textural properties of the material. Ammonia temperature-programmed-desorption (NH3-TPD) measurements and infrared spectra using probe molecules such as pyridine (Py-IR) and 2,4,6-collidine (Coll-IR) have been collected to investigate the acidic properties as well as the accessibility of the acid sites. The hierarchical ZSM-11 possesses more acid sites on the mesopore surfaces that are accessible towards large probe molecules such as 2,4,6-collidine. This improvement together with the enhanced pore-connectivity brings about an increase in 1,3,5-triisopropylbenzene cracking activity and benzene selectivity with respect to a conventional counterpart.
1. Introduction
Zeolites are a family of microporous (<2 nm) crystalline solids with ordered pore size, shape and connectivity that impart them with unique size and shape selectivity to guest molecules that can be accommodated in their pores.1,2 Artificial zeolites are nowadays mass produced on an industrial scale and find broad applications in areas such as ion-exchangers in laundry, adsorbents for separation, and more importantly as solid acid catalysts in oil refinery and chemical industries.1,3 Moreover, zeolites are also the preferred solid acid catalysts for a series of important transformations using coal, natural gas or biomass as feedstocks.4–7 Nowadays, the frequently used artificial zeolites are micronsized crystals, whereas their pore sizes are no more than 2.0 nm. Consequently, the mass transport in the micropores of zeolites are restricted, which undermines the effectiveness and fields of applications for zeolites.8–12 In the past 15 years or so, two major strategies have been used to facilitate mass transport in zeolitic micropores: (1) downsize the crystal of zeolites from microns to nanometers, viz., synthesizing nanozeolites13–15 or zeolites with plate-like morphology of nanometer thickness.16–19 (2) Introducing auxiliary meso- or macro-pores in addition to the inherent micropores of zeolites, i.e., generating hierarchically porous zeolites (HPZs) to take the place of conventional zeolites that possess solely micropores.20–24 Hard template route using carbon blacks,25 soft template route using organosilanes,26–31 cationic polymers,32 bifunctional structure directing agents,18,33,34 as well as postsynthetic desilication35,36 routes have been established. Several merits stems from prompted mass transport for instance, high activity, occasional high primary product selectivity, improved large molecule accessibility, as well as longer lifetimes, have been achieved by replacing conventional zeolites with HPZs.37 More recently, there are several reports on the industrial applications for this type of zeolitic catalysts,35,38,39 and more examples are expected to come in the future.From the mass transport viewpoint, molecular transport undergoes configurational diffusion in the micropores of zeolites, which can be understood by taking into the Thiele modulus (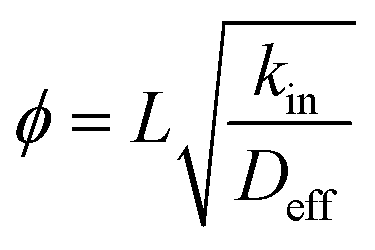 , whereby L, kin, Deff stands for diffusion length, intrinsic reaction rate constant and effective diffusivity, respectively).8 The larger the Thiele modulus, the more serious the effect of intra-particle diffusion over reaction. Advantages of nanozeolites or introducing auxiliary porosity originate from the fact that the diffusion pathway (L) has been shortened, henceforth, the utility of zeolitic material is enhanced.6 On the other hand, it has been recognized that the extent to which mass transport is improved is highly dependent on the “quality” of meso- (or macro-)pores, as mesopores can enhance mass transport only if they are connected with the exterior surface of zeolite crystals.36,38,40 Weitkamp et al.41 have observed that the presence of intracrystal mesopores in USY has virtually no impact on mass transport, a phenomenon that has also been recently corroborated by Perez-Ramirez et al. using positron annihilation lifetime spectroscopy.36,38 It is therefore desirable to find ways that can produce HPZs with small primary particle size that can shorten the diffusion pathway, and at the same time keep the mesopores connected to the external surfaces. ZSM-11 possesses a MEL topology that is an important member among the pentasil family of zeolites, the MEL structure consists of intersecting straight 10 member ring channels (5.3 × 5.4 Å) that is similar to ZSM-5 (MFI topology).42 The first mesoporous ZSM-11 (Si/Al = 50) has been obtained by Christensen et al.25 using carbon black as hard template, the Cu-containing mesoporous ZSM-11 is more catalytically active than the conventional counterpart or the mesoporous ZSM-5 in NO decomposition. The improved accessibility in the straight channels compared to the sinusoidal channels together with the presence of auxiliary mesopores are responsible for the elevation of activity.43 Xie et al.44 have reported the synthesis of mesoporous ZSM-11 (Si/Al = 19) through polyvinyl butyral (PVB) templating method, and the mesoporous ZSM-11 overperforms microporous ZSM-11 in cracking of both 1,2,4-trimethylbenzene and 1,3,5-trimethylbenzene. The generation of mesoporous ZSM-11 has also been reported by using cetyltrimethylammonium tosylate and TBAOH in a dual-templating synthetic system by a two-step preparation,45 but the competing role between the two structure directing agents and consequently the phase segregation can hardly be ruled out as found for similar synthetic systems.24,46
, whereby L, kin, Deff stands for diffusion length, intrinsic reaction rate constant and effective diffusivity, respectively).8 The larger the Thiele modulus, the more serious the effect of intra-particle diffusion over reaction. Advantages of nanozeolites or introducing auxiliary porosity originate from the fact that the diffusion pathway (L) has been shortened, henceforth, the utility of zeolitic material is enhanced.6 On the other hand, it has been recognized that the extent to which mass transport is improved is highly dependent on the “quality” of meso- (or macro-)pores, as mesopores can enhance mass transport only if they are connected with the exterior surface of zeolite crystals.36,38,40 Weitkamp et al.41 have observed that the presence of intracrystal mesopores in USY has virtually no impact on mass transport, a phenomenon that has also been recently corroborated by Perez-Ramirez et al. using positron annihilation lifetime spectroscopy.36,38 It is therefore desirable to find ways that can produce HPZs with small primary particle size that can shorten the diffusion pathway, and at the same time keep the mesopores connected to the external surfaces. ZSM-11 possesses a MEL topology that is an important member among the pentasil family of zeolites, the MEL structure consists of intersecting straight 10 member ring channels (5.3 × 5.4 Å) that is similar to ZSM-5 (MFI topology).42 The first mesoporous ZSM-11 (Si/Al = 50) has been obtained by Christensen et al.25 using carbon black as hard template, the Cu-containing mesoporous ZSM-11 is more catalytically active than the conventional counterpart or the mesoporous ZSM-5 in NO decomposition. The improved accessibility in the straight channels compared to the sinusoidal channels together with the presence of auxiliary mesopores are responsible for the elevation of activity.43 Xie et al.44 have reported the synthesis of mesoporous ZSM-11 (Si/Al = 19) through polyvinyl butyral (PVB) templating method, and the mesoporous ZSM-11 overperforms microporous ZSM-11 in cracking of both 1,2,4-trimethylbenzene and 1,3,5-trimethylbenzene. The generation of mesoporous ZSM-11 has also been reported by using cetyltrimethylammonium tosylate and TBAOH in a dual-templating synthetic system by a two-step preparation,45 but the competing role between the two structure directing agents and consequently the phase segregation can hardly be ruled out as found for similar synthetic systems.24,46
In preceding articles, we have used an organosilane and dry gel conversion combined strategy to generate HPZs.29–31 The organosilane route has been initiated by Serrano et al.27,47,48 as an effective route to control zeolite crystal size, but entails surface modification of zeolitic embryos before a second crystallization process. The dry gel conversion route has been proposed by Xu et al.49 and later developed by Matsukata et al.,50,51 Davis,52 and many others into an alternative route for zeolites or zeotype material synthesis.2 By combining the two approaches, it is possible to control the texture of the dry gel and avoid the fusion of primary zeolitic particles into large crystals. Meanwhile, the phase separation of organosilanes from zeolite crystals is inhibited, that has been found to be important to the creation of mesopores.30,47,53 Herein, we report the synthesis of HPZ ZSM-11, by using solvent evaporation route to generate a preformed dry gel and its subsequent steam-assisted-crystallization (SAC) treatment to convert the dry gel into zeolites. The primary crystal size has been found to be as small as ca. 35 to 60 nm and pore quality is high. The acidity and accessibility has been measured and catalytic performance has been probed using 1,3,5-triisopropylbenzene cracking as a model reaction.
2. Experimental section
2.1. Reagents
Hexadecyltrimethoxysilane (HTS, 85 wt%, Aladdin), aluminum tert-butoxide (ATTB, 99 wt%, J&K Chemical), tetrabutylammonium hydroxide (TBAOH, 25 wt% in water, Aladdin), ethyl silicate (TEOS, 28 wt% SiO2, Shanghai Lingfeng Chemical Reagent Co., Ltd), ethanol (99.7%, Shanghai Titan Scientific Co., Ltd), chloroplatinic acid hexahydrate (H2PtCl6·6H2O, 37.5 wt% Pt, Aladdin), hydrogen fluoride solution (HF, 40.6 wt%, Shanghai Lingfeng Chemical Reagent Co., Ltd) were of analytical reagent grade and used without further purification.2.2. Synthesis
The molar composition of the starting gel used for the synthesis of hierarchical porous zeolite ZSM-11 was: 0.95SiO2/0.05HTS/0.01Al2O3/0.20TBAOH/8.65H2O. In a typical synthesis, 0.29 g of ATTB was dissolved in 10.39 g of TBAOH aqueous solution followed by adding 100 ml of ethanol at room temperature under stirring for 30 min to form an apparent clear mixture. Another clear mixture of 10.72 g of TEOS, 1.06 g of HTS and 50.0 ml of ethanol was made by vigorously stirring for 2 h. The above two mixtures were mixed and stirred for another 2 h to afford a clear solution-like mixture. The mixture was transferred into a Petri dish to allow the evaporation of solvent that produced a transparent dry gel. The dry gel was ground to powder by mortar and pestle before charging into a 10 ml Teflon cup placed inside Teflon-lined stainless steel autoclave (100 cm3), and 5.0 g H2O was added outside the cup to create enough steam for SAC treatment. Then the autoclave was kept in an oven to carry out a SAC type treatment at 453 K up to 4 days. The product was recovered by filtration, washing thoroughly with deionized water, and dried at 373 K for 5 h. The organic template was removed by calcination for 20 h in air with a ramp of 2 K min−1 to 823 K. Thus the final product named ZSM-11-H was produced. For comparison, conventional ZSM-11 was synthesized by the same procedure in the absence of HTS, and the starting gel composition of conventional porous zeolite ZSM-11 was: 1.00SiO2/0.01Al2O3/0.20TBAOH/8.65H2O. The final obtained sample was labeled as ZSM-11-C, whereby C stands for conventional microporous sample.Pt replica synthesis: 0.5 g calcined ZSM-11-H was mixed with 0.30 g H2PtCl6·6H2O in a glass beaker, where it was kept stirring at 363 K for 8 h, subsequently, the solid was obtained by centrifugation and then heated at 673 K in H2 for 2 h. The zeolite was dissolved by a hydrogen fluoride solution (HF), and the Pt product was repeatedly washed with water and ethanol.
2.3. Characterizations
Powder X-ray diffraction (XRD) patterns of samples were recorded at a Rigaku D/Max 2550VB/PC powder diffractometer with Cu Kα radiation (λ = 1.5418 Å) at 40 kV and 100 mA. Scanning electron micrographic (SEM) and transmission electron micrographic (TEM) images were obtained on a Nova NanoSEM450 microscope and a JEM-2100 microscope, respectively. To prepare the sample for TEM, the powder was suspended in ethanol and supported onto a lacey carbon film on a copper grid. Nitrogen adsorption–desorption isotherms were measured on Micromeritics ASAP 2020 analyzer at 77 K. Before measurements, calcined samples were degassed at 553 K for 10 h. The specific surface areas (SBET) were calculated by the multi-point Brunauer–Emmett–Teller (BET) method. The external specific surface area and micropore pore volume were evaluated by the t-plot method. The pore size distribution was estimated by applying the NL-DFT (Non-Local Density Functional Theory) method to the adsorption branches of the isotherms, and the total pore volumes were estimated from the adsorbed amount at a relative pressure P/P0 = 0.99. Ammonia temperature programmed desorption (NH3-TPD) were performed on PX200A type TPD/TPR setup. A sample of 100 mg was activated in helium flow at 873 K for 1 h and then cooled to 373 K for the saturation of NH3. The NH3-TPD profile was recorded by heating the sample at a rate of 15 K min−1 from 423 K to 923 K under a helium flow (30 cm3 min−1). Transmission FTIR spectra of the samples were recorded with a Spectrum 100 FT-IR spectrometer. All samples were pressed in self-supporting wafers (diameter: 1.4 cm, weight: 166 mg) and were pre-treated at 773 K under vacuum for 1 h prior to the adsorption of probe molecules (pyridine or 2,4,6-collidine). The amount of adsorbed probe molecules was calculated from the integrated area of given bands with their distinct molar extinction coefficients. For 2,4,6-collidine, εH-bond (1632–1648 cm−1) = 10.1 cm μmol−1 has been used to calculated the amount of acid sites that resides on the mesopore wall or external surface of zeolites.54 For pyridine, εBrønsted (1540 cm−1) = 1.02 cm μmol−1 and εLewis (1450 cm−1) = 0.89 cm μmol−1 has been employed to deduce the total amount of Brønsted and Lewis acid sites, respectively, that are given in literature.542.4. Catalytic assessments
The 1,3,5-triisopropylbenzene cracking was chosen as a model reaction because the substrate 1,3,5-triisopropylbenzene has a larger dynamic size (9.5 Å)44 than the micropore pore size of ZSM-11 (5.3 × 5.4 Å). The cracking reaction was carried out on a pulse micro-reactor. 0.1 g catalyst (10–20 mesh) was packed between quartz wool plugs in a U-shaped stainless tube (i.d. = 4 mm) in a furnace. About 0.4 μL of reactant was injected every time. The products were analyzed using Gas Chromatograph (GC) with a capillary column of OV-101 and a flame ionization detector (FID).3. Results and discussion
3.1. Crystal and texture properties of ZSM-11-H
The structure of the calcined ZSM-11-H obtained after solvent evaporation and SAC treatment has been characterized by a combination of various techniques. Fig. 1 shows the powder XRD patterns of ZSM-11-C and ZSM-11-H. The feature XRD reflections of ZSM-11 are at 2θ of 7.92, 8.78, 23.14, 23.98, and 45.2°, together with double peaks at 23–25° and the single peak at 45° are used to distinguish MEL from that of MFI structures.25,42,44 Both samples exhibit reflections typical of the MEL topology and no impurities have been detected, denoting an absence of amorphous gel or other phases. In addition, broadening of diffraction peaks for ZSM-11-H is intuitively seen which is ascribed to the smaller particle size respect to the conventional hydrothermal method. The inference of particle size is not possible by the XRD techniques using the Scherrer equation,55 because of the intergrowth nature of the material (see later). | ||
| Fig. 1 XRD patterns of ZSM-11-C and ZSM-11-H. | ||
N2 adsorption–desorption isotherms have been measured to provide the texture properties of ZSM-11-H, that is juxtaposed together with the data for ZSM-11-C for comparison, as exhibited in Fig. 2 and the derived data tabulated in Table 1. The isotherm for ZSM-11-C can be ascribed to a type I according to the IUPAC classifications, the inherent micropores in zeolite has been filled at relatively low pressures (P/P0 < 0.1). At P/P0 above 0.95, a small jump in N2 uptake is observable, that is due to capillary effect caused by the presence of interparticular voids. The isotherm for ZSM-11-H is a hybrid type I and IV isotherm, besides the micropore filling at low relative pressures (P/P0 < 0.1), the isotherm contains an addition N2 uptake at P/P0 > 0.80, which can be attributed to the capillary effect caused by the presence of mesopores.25,44 The corresponding hysteresis loop can be categorized to a type H3, implying an irregularly shaped mesopore. The absence of a forced closure in the hysteresis loop of the isotherm at P/P0 ≈ 0.45 caused by tensile-strength-effect (TSE) indicated the formation of accessible mesoporosity.56,57 As shown in Table 1, the micropore volumes for the two samples are comparable, suggesting that the introduction of mesopores has negligible effect on the inherent microporosity of MEL structure. The external surface area and mesopore volume for ZSM-11-C are 160 m2 g−1 and 0.11 cm3 g−1 due to surface roughness and exterior surface, respectively, whereas for ZSM-11-H a high external surface area (252 m2 g−1) and mesopore volume (0.54 cm3 g−1) can be derived. It can be deduced that the mesopore volume is attributed to the aggregation of primary particulates, which also lead to the increase of surface area and mesopore volumes. A SBET of 363 m2 g−1 and Langmuir surface area of 611 m2 g−1 is derived for ZSM-11-H, that is larger than that for ZSM-11-C (SBET = 317 m2 g−1, SLangmuir = 556 m2 g−1). Considering the different porosity of ZSM-11-C and ZSM-11-H, one can conclude that the addition of HTS by solvent evaporation route has created mesoporous region in the material by expanding interparticular spacing between primary particles. It is noteworthy that the mesopore volume is larger than hierarchical ZSM-11 derived from either hard template route (0.40 m2 g−1)25 or PVB template (0.23 m2 g−1).44
 | ||
| Fig. 2 N2 adsorption–desorption isotherm of ZSM-11-C and ZSM-11-H, and the corresponding pore-size-distribution pattern for ZSM-11-H sample was shown in the inset. | ||
| Sample | SBETa (m2 g−1) | SLangmuirb (m2 g−1) | Sextc (m2 g−1) | Vtotal (cm3 g−1) | Vmicroc (cm3 g−1) | Vmesod (cm3 g−1) |
|---|---|---|---|---|---|---|
| a Calculated by the BET method in the P/P0 range of 0.05–0.20.b Calculated using the Langmiur method.c Calculated using the t-plot method.d Calculated using Vtotal − Vmicro. | ||||||
| ZSM-11-C | 317 | 556 | 160 | 0.17 | 0.12 | 0.05 |
| ZSM-11-H | 363 | 611 | 252 | 0.65 | 0.11 | 0.54 |
SEM images have been collected to throw light on the effect of organosilane HTS over the morphology of ZSM-11 zeolites. The SEM images of ZSM-11-H and ZSM-11-C are displayed in Fig. 3. The ZSM-11-C has typical olive-like shape of MEL zeolite crystallites with the smooth surface and clear crystalline edges and compact texture, indicating the solely presence of inherent micropores in the sample.25 And the average particle size of ZSM-11-C is approximately 450–500 nm as can be detected in the SEM images at low magnifications. In contrast, the crystal size of ZSM-11-H lays in the range of 380–480 nm, and each discrete crystal is made up of tiny primary particles between 35 to 60 nm. The crystal size for ZSM-11-H is smaller than that of hierarchical ZSM-5 reported previously,29 presumably due to the differences in crystalline structure and the structure-directing-agents (SDAs). The surface of ZSM-11-H appears rough and sponge-like, that is similar to other hierarchical zeolites and strongly indicating the presence of additional porosity within the crystal.25,33,44,46,58
 | ||
| Fig. 3 SEM images for calcined samples, ZSM-11-C (a and b) and ZSM-11-H (c and d). | ||
TEM images of the ZSM-11-H are shown in Fig. 4. ZSM-11-H exhibits crystalline edges of MEL structure in some area with fluffy-like morphology, mesopore ranges appearing as white areas can be visualized between the primary crystal particles. The white fields as mesopores are randomly distributed throughout the whole crystal, as a result of aggregation of primary building blocks. Another feature is that the mesopores are not intracrystal, but are connected to the external surfaces. The TEM images also reveal that olive-like spheres ZSM-11-H particles are composed of large numbers of nanosize zeolite (from 35 to 60 nm), according well with the SEM results. The selected area electron diffraction (SAED, in the inset Fig. 4b) pattern for ZSM-11-H shows that each crystal possesses single-crystal-like diffraction spots, confirming that the primary zeolite particles are mutually aligned. The structure is therefore similar to the diffraction patterns for mesocrystals, whereby nanoparticles form superstructures when they are orientated along the same crystalline axis.59 The lattice fringes could be clearly distinguished at high magnification (Fig. 4d), showing that zeolite ZSM-11-H is highly crystalline. The alignment of these primary particulates are similar to ZSM-5 derived from solvent-evaporation and dry gel conversion protocol,29 zeolite beta with similar formation mechanism has also been unraveled recently.46 The ordered micropores and channels arisen from the crystalline structure are well arranged in the crystal.
 | ||
| Fig. 4 TEM images for ZSM-11-H after calcinations, the SAED pattern was shown in the insets. | ||
To shed light on the mesopore connectivity, a nanocasting approach has been used to prepare a Pt particle that is a replica of the mesopores for ZSM-11-H, the TEM image for this Pt particle is exhibited in Fig. 5. Pt derived from nanocasting using silica as a hard template has been reported before by Fan et al.,60 as the Pt-replica honestly preserves the shape of the pores of parent template, the Pt-replica can be employed to estimate the pore connectivity for the parent hard template (mold). It is seen from Fig. 5 that the Pt-replica appear as dark dense solids of overlapping small particles of 2–5 nm, constituting aggregates of varied bigger sizes, which is consistent with the broad mesopore size distribution deduced from N2 physisorption data. Most of the aggregated areas are too thick to provide information on pore-connectivity, but on the margins of some areas, worm-like shaped Pt-replicas can be identified (as specified by arrows), suggesting that the mesopores are worm-like in shape and are interconnected to construct a porous system.
 | ||
| Fig. 5 TEM image for Pt-replica derived from ZSM-11-H by a nanocasting approach. | ||
3.2. Formation process of ZSM-11-C and ZSM-11-H
The time-dependent structure evolution during steaming treatment has been monitored by XRD and SEM for quenched samples collected at varied time intervals for both ZSM-11-C and ZSM-11-H, these results are displayed in Fig. 6 and 7, with the intention to understand the crystallization process. For ZSM-11-C, the initial dry gel after solvent evaporation is amorphous in nature, as exhibited by the lump diffraction pattern from 18 to 30° (Fig. 6a) and the corresponding SEM image (Fig. 7a). The dry gel comprises of partly fused particulates between 9 and 15 nm, with occasional bigger ones that can be also be found in the sample. After 6 h of SAC treatment, XRD detectable transformation from dry gel to MEL structure takes place, when a noticeable diffraction out of MEL structure is observable. The gel has decomposed into small particle of 10–20 nm aggregates. Prolonged crystallization for 12 h leads to the formation of more MEL phase and fusion of these tiny zeolitic embryo into bulky crystals. At this stage, particles with an average size of 10–30 nm can be identified, whereby grain boundaries are seen more clearly due to ordering development. For the same sample, it is found that attachment of primary particles into agglomerates has occurred in most areas. After 48 h, majority of the starting gel has been converted into MEL zeolites, when no amorphous diffraction patterns can be identified, and the typical round shape of 300–700 nm MEL crystal is clearly visible. The disappearance of grain boundaries between primary particulates can be visualized as well. The temporal overlap of crystallization and morphology change from tiny nanocrystals to aligned bulky crystal is related to the crystal growth mechanism, which is frequently found for growth of crystals undergoing an orientated attachment (OA) route.61,62 The formation and alignment of zeolitic crystals has been proposed as a result of OA growth by Bein et al.,58 Matsukata et al.51 and us46 in the case of beta zeolite formation in dry gel system, similar phenomenon can occur to ZSM-11. Tsapatsis et al.63 have also proposed that siliclite-1 (MFI) growth in solution system also takes place via attachment of primary building units. It is speculated that OA growth can be the dominant route in dry gel systems, as supersaturation ratio is high and less water is available for the complete dissolving of precursor ingredients. The high supersaturation ratio guarantees a high nucleation rate that lead to the formation of large numbers of nucleates at the early stage of crystallization, while the lack of water does not favor the transport of starting materials and further growth happens mainly through the alignment of primary crystals. Based on the above observations, a possible formation mechanism has been sketched in Scheme 1a. | ||
| Fig. 6 Time-dependent XRD patterns for dry gel after steaming treatment at varied time in the synthesis of ZSM-11-C (a) and ZSM-11-H (b). | ||
 | ||
| Fig. 7 The SEM images for dry gel after steaming treatment for varied time in the synthesis of ZSM-11-C (a) and ZSM-11-H (b). | ||
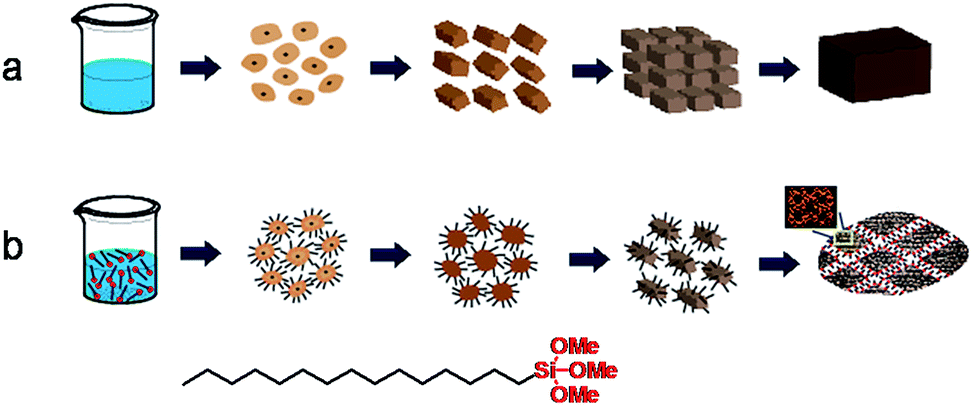 | ||
| Scheme 1 The proposed formation mechanism for ZSM-11-C (a) and ZSM-11-H (b) in solvent evaporation and steam-assisted-crystallization process. | ||
For ZSM-11-H, the dry gel after solvent evaporation appears to be amorphous (Fig. 6b) and more uniform in size by the inclusion of HTS, and a particle size between 10 to 20 nm can be inferred from SEM images (Fig. 7b). The gel becomes aggregates made up of 15–30 nm particulates after 6 h of SAC treatment and remains amorphous. The crystallization starts only after 12 h SAC treatment, i.e., the crystallization has been slowed down as inhibited by HTS. Even after 48 h of SAC treatment, an obvious broad diffraction out of amorphous gel can be found, indicating that the majority of the starting gel has not been transformed. For the 48 h treated sample, the gel comprises particulates between 35–50 nm with open framework structures that is rich in porous region. Compared with the final products (Fig. 3c and d), it is obvious that the ZSM-11-H has grown from these discrete particulates, the crystallization of MEL structure is accompanied with prominent morphology change at the mesoscale. The role of HTS can be understood by comparing with the crystallization of ZSM-11-C. HTS slows down the rate of crystallization in a OA growth mechanism, meanwhile, the aggregation of primary particulates into bulky crystals has been inhibited. The alkyl tails of HTS form the interparticular voids that form mesopores during SAC treatment. This process has been depicted in Scheme 1b to highlight the role of HTS.
3.3. Acid properties and catalytic performance
Fig. 8 presents the NH3-TPD profiles of the ZSM-11-C and ZSM-11-H. The acid concentrations of differing strengths derived from the profiles are formulated in the Table 2. Obviously, there are two desorption peaks for the two samples, corresponding to the weak and strong acid sites, respectively. The low temperature desorption of NH3 is attributed to the physically adsorbed NH3, while the high temperature desorption peak is associated with NH3 interacting with strong acid sites.44,64 The high temperatures of desorption for ZSM-11-C and ZSM-11-H appear at 682 and 676 K, the close in desorption temperature shows that the acidic strength for the two samples are comparable, i.e., the integration of mesopores to the native micropores in MEL structure by our method does not cause substantial decrease in strong acidic strength. From the Table 2, it can be estimated that the number of strong acid sites for ZSM-11-C and ZSM-11-H are 0.58 and 0.62 mmol g−1-cat., implying that the two samples have similar numbers for strong acid sites. The ZSM-11-H possesses less weak acid site (0.24 mmol g−1-cat.) and more strong acid site (0.62 mmol g−1-cat.), that is different from previously reported hierarchical ZSM-11 templated by PVB.44 | ||
| Fig. 8 NH3-TPD profiles of ZSM-11-C and ZSM-11-H. | ||
FT-IR measurements were employed to distinguish the acid type and the acidic accessibility of those samples by using adsorbed pyridine (Py, 5.7 Å in diameter) and 2,4,6-collidine (Coll, 7.4 Å in diameter) as probe molecules. As illustrated in Fig. 9a, the adsorption bands at 1454 cm−1 and 1542 cm−1 are assigned to the chemisorbed pyridine, which correspond to the pyridine interacting with Lewis and Brønsted acid sites, respectively.65 The peak at 1490 cm−1 is synergetic result from both Lewis and Brønsted acid sites.65 The areas of the peaks decrease gradually with the raising of outgassing temperatures from 473 K to 673 K, as a consequence of weakly adsorbed pyridine desorption at elevated temperatures. The Py-IR and Coll-IR results have been analyzed to generate information on the type and accessibility of acid sites,57 with respect to previously established methods. The detailed results are compiled in the Table 3. Despite the fact that the total acid sites are similar for ZSM-11-C (0.64 mmol Py g−1-cat.) and ZSM-11-H (0.56 mmol Py g−1-cat.), the number for strong Brønsted acid sites (measured at 673 K) are 0.26 and 0.10 mmol Py g−1-cat., respectively. Meanwhile, the measured numbers for strong Lewis acid sites (measured at 673 K) are 0.01 and 0.11 mmol Py g−1-cat., for ZSM-11-C and ZSM-11-H, respectively. The Py-IR data point out that the integrating of auxiliary porosity using our method has altered the type of strong acid sites from a Brønsted-acid-sites-rich type to a Lewis-acid-sites-rich type. The International Zeolite Association (IZA) shows that the channel diameter of ZSM-11 zeolite is about 5.3 × 5.4 Å, which is similar to the dynamic molecular size of pyridine (5.7 Å (ref. 57)), and pyridine can enter the micropores of MEL structure.66,67 Therefore, the Py-IR results reflect the overall acid properties without discriminating acid sites located on zeolite external surfaces or their internal micro-channels. 2,4,6-collidine, on the other hand, is bulkier (7.4 Å) than pyridine and can only access to Brønsted acid sites through larger auxiliary pores,57 that could only be used to probe acid sites situated at auxiliary mesopores and the external surfaces of MEL structure. As the intracrystalline mesopores can only be accessed through the micropores, whereby the 2,4,6-collidine adsorption is normally denied, the Coll-IR is henceforth speculated to only give the number of acid sites sitting on mesopores that are accessible from exterior surface of zeolite crystals.36 The vibrations at 1638 and 1633 cm−1 have been assigned to 2,4,6-collidine molecule interacting with Brønsted and Lewis acid sites according to Gilson et al.,57,68 and only 0.04 mmol g−1-cat. is measurable on the exterior surface of ZSM-11-C. As shown in Table 3, it can be noticed that the 2,4,6-collidine adsorption on the hierarchical zeolite ZSM-11-H (0.10 mmol Coll g−1-cat.) is obviously higher than that of conventional zeolite ZSM-11-C (0.04 mmol Coll g−1-cat.), and the ratio for 2,4,6-collidine accessible acid sites on ZSM-11-H to ZSM-11-C at 673 K is 2.5. This result evidences that the appearance of mesopores in ZSM-11-H zeolite leads to the increment of available acid sites for bulky molecules. This observation is in accordance with the reported trend that mesopore surface area increases hand-in-hand with the accessibility of acidic sites.57 From the above acid property measurements, it is concluded that ZSM-11-H has similar total strong acid sites to that of ZSM-11-C. The bulky molecule accessibility to acid sites has been improved with the integration of auxiliary mesopores. There are more Lewis acids sites for ZSM-11-H than on ZSM-11-C, as can be inferred from pyridine IR data in Table 3.
 | ||
| Fig. 9 Pyridine (a) and 2,4,6-collidine (b) infrared spectra for ZSM-11-C and ZSM-11-H. | ||
| Sample | Total aciditya mmol g−1 | Aciditya (mmol Py g−1-cat.) | Acidityb (mmol Coll g−1-cat.) (1632–1648 cm−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Brønsted (1554 cm−1) | Lewis (1452 cm−1) | |||||||||
| 473 K | 573 K | 673 K | 473 K | 573 K | 673 K | 473 K | 573 K | 673 K | ||
| a Calculated by Py-IR.b Calculated by Coll-IR. | ||||||||||
| ZSM-11-C | 0.64 | 0.60 | 0.42 | 0.26 | 0.04 | 0.03 | 0.01 | 0.09 | 0.07 | 0.04 |
| ZSM-11-H | 0.56 | 0.42 | 0.15 | 0.10 | 0.14 | 0.13 | 0.11 | 0.21 | 0.11 | 0.10 |
The ultimate goal of fabricating hierarchical zeolite is to promote the catalytic performances, especially the accessibility and diffusion pathway for bulky reactants. The catalytic activity of ZSM-11-H has been investigated in 1,3,5-triisopropylbenzene (1,3,5-TIPB) cracking at 623 and 673 K, respectively. 1,3,5-TIPB is regarded as a model compound for heavy gas oil molecules, and is widely accepted as a probe reaction to estimate the effect of hierarchical porosity of the catalytic performance. The catalytic results obtained over the two catalysts at 623 and 673 K are presented in Table 4. The 1,3,5-TIPB conversion for conventional ZSM-11-C is 51.9% and 60.3% at 623 K and 673 K, respectively. Interestingly, the 1,3,5-TIPB conversion over hierarchical ZSM-11-H is 68.2% and 77.1% at 623 K and 673 K, respectively. This is because the parent ZSM-11-C has only native micropore diameter no larger than 5.6 Å, that is significantly smaller than the kinetic diameter of 1,3,5-TIPB (9.5 Å) and thus their catalytic conversion occurs exclusively at the exterior surface of ZSM-11 crystal. The small external surface area (Table 1) and the sterically hindered accessibility to acid sites (as probed by 2,4,6-collidine) strongly limits the effectiveness of ZSM-11-C for the reaction, consequently, the catalytic reaction only occurs on the external surfaces and pore-mouth of the ZSM-11-C crystals. On the other hand, for ZSM-11-H, the presence of mesopores together with the improved pore connectivity, enhanced bulky molecule accessibility towards catalytic sites offer the opportunity of more available sites to participate the catalysis, leading to an increase in conversion. The primary product after 1,3,5-triisopropylbenzene de-alkylation is 1,3-diisopropylbenzene (1,3-DIPB, 8.4 Å), that in turn, can be de-alkylated to cumene (6.8 Å) or further to produce benzene, with propylene as a co-product in each consecutive step. These molecules are small enough to penetrate the micropores system to undergo further cracking at reaction conditions, henceforth, the products distribution reflect the effect out of diffusion rather than accessibility.69,70 In ZSM-11-H, as the diffusion pathway has been shortened as the crystal size is reduced, more primary product 1,3-diisopropylbenzene has been converted into cumene and benzene, resulting in a detectable increase in their selectivities (Table 4). It should be noted that the selectivity to tetraisopropylbenzenes (TetraIPBs) is much higher over ZSM-11-H at 623 K. This is probably due to the disproportionation reaction of 1,3,5-triisopropylbenzenes, or the alkylation reaction between 1,3,5-triisopropylbenzene and propylene. The formation of more tetraisopropylbenzenes further proves the improved accessibility of ZSM-11-H. As the temperature rises the selectivity to tetraisopropylbenzene decreases from 15.1 to 3.4 over ZSM-11-H. This is because that high temperature is conducive to cracking reaction, which leads to the cracking of tetraisopropylbenzene and the formation of more benzene and cumene. The effect of hierarchical porous structure on the cracking of 1,3,5-TIPB molecule is an increase in activity stemming from more accessible acid sites, and an increased selectivity towards consecutive de-alkylation products due to reduced diffusion length in micropores.
| Sample | T (K) | Conv. (%) | Production distribution (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Propylene | Benzene | Cumene | 1,3-DIPB | 1,4-DIPB | TetraIPBs | |||
| ZSM-11-C | 623 | 51.9 | 32.7 | 21.6 | Trace | 42.7 | Trace | 3.0 |
| ZSM-11-H | 623 | 68.2 | 28.9 | 23.9 | 6.1 | 26.0 | Trace | 15.1 |
| ZSM-11-C | 673 | 60.3 | 36.4 | 24.5 | Trace | 34.1 | Trace | 5.0 |
| ZSM-11-H | 673 | 77.1 | 39.6 | 29.5 | 7.8 | 19.7 | Trace | 3.4 |
4. Conclusions
A new route to generate hierarchically porous ZSM-11 has been paved by a organosilane assisted solvent evaporation route to fabricate dry gel and its subsequent transformation into zeolites. The hierarchical ZSM-11-H possesses both native micropores and auxiliary mesopores, where mesopores are interconnected. The synthesis merits small primary particle size and well mesopore-connectivity. The introduction of mesopore also lead to the formation of more Lewis acid sites than Brønsted sites, while the total number of strong acid sites are almost identical to that of microporous ZSM-11. Meanwhile, the accessibility to the otherwise sterically hindered Brønsted sites has been enhanced. ZSM-11-H shows improved catalytic activity in probe catalytic assessment for 1,3,5-triisopropylbenzene cracking reaction with respect to the conventional ZSM-11.It is observed that orientated attachment growth can be the dominating crystallization route for ZSM-11 growth in a dry gel system, thereby the method can be extrapolated to other dry gel systems. Applicability of such a strategy in synthesizing other zeolites or zeotype materials is underway and will be reported in the future.
Acknowledgements
KZ is grateful for the financial support from Fundamental Research Funds for the Central Universities (WB 1213004-1), and New Century Excellent Talents in University (NCET-11-0644). KZ is also indebted to ELECTRONANOMAT (Grant Agreement Number: PIRSES-GA-2012-318990) sponsored by the Marie-Curie Act program of EU for the trip to Denmark. XZ is thankful for National Natural Science Foundation of China (U1162112) for the financial support. ZL is grateful for the Fundamental Research Funds for the Central Universities (WA 1514011).References
- C. Martinez and A. Corma, Coord. Chem. Rev., 2011, 255, 1558–1580 CrossRef CAS PubMed
.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–701 CrossRef CAS PubMed
- M. Moliner, C. Martinez and A. Corma, Chem. Mater., 2014, 26, 246–258 CrossRef CAS
- U. V. Mentzel, S. Shunmugavel, S. L. Hruby, C. H. Christensen and M. S. Holm, J. Am. Chem. Soc., 2009, 131, 17009–17013 CrossRef CAS PubMed
- W. Kim and R. Ryoo, Catal. Lett., 2014, 144, 1164–1169 CrossRef CAS PubMed
- J. Mielby, J. O. Abildstrom, F. Wang, T. Kasama, C. Weidenthaler and S. Kegnaes, Angew. Chem., Int. Ed., 2014, 53, 12513–12516 CAS
- K. T. Hojholt, A. B. Laursen, S. Kegnaes and C. H. Christensen, Top. Catal., 2011, 54, 1026–1033 CrossRef CAS
- C. H. Christensen, K. Johannsen, E. Toernqvist, I. Schmidt, H. Topsoe and C. H. Christensen, Catal. Today, 2007, 128, 117–122 CrossRef CAS PubMed
- C. H. Christensen, K. Johannsen, I. Schmidt and C. H. Christensen, J. Am. Chem. Soc., 2003, 125, 13370–13371 CrossRef CAS PubMed
- O. C. Gobin, S. J. Reitmeier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2009, 113, 20435–20444 CAS
- M. R. Bonilla, T. Titze, F. Schmidt, D. Mehlhorn, C. Chmelik, R. Valiullin, S. K. Bhatia, S. Kaskel, R. Ryoo and J. Kaerger, Materials, 2013, 6, 2662–2688 CrossRef CAS PubMed
- K. Cho, H. S. Cho, L. C. D. Menorval and R. Ryoo, Chem. Mater., 2009, 21, 5664–5673 CrossRef CAS
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734–6760 CrossRef CAS PubMed
- N. Eng-Poh, D. Chateigner, T. Bein, V. Valtchev and S. Mintova, Science, 2012, 335, 70–73 CrossRef PubMed
- M. A. Camblor, A. Corma and S. Valencia, Microporous Mesoporous Mater., 1998, 25, 59–74 CrossRef CAS
- L. Wang, Y. Wang, Y. Liu, L. Chen, S. Cheng, G. Gao, M. He and P. Wu, Microporous Mesoporous Mater., 2008, 113, 435–444 CrossRef CAS PubMed
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 CrossRef CAS PubMed
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684–1687 CrossRef CAS PubMed
- E. Verheyen, L. Joos, K. Van Havenbergh, E. Breynaert, N. Kasian, E. Gobechiya, K. Houthoofd, C. Martineau, M. Hinterstein, F. Taulelle, V. V. Speybroeck, M. Waroquier, S. Bals, G. V. Tendeloo, C. E. A. Kirschhock and J. A. Martens, Nat. Mater., 2012, 11, 1059–1064 CAS
- K. Moeller and T. Bein, Chem. Soc. Rev., 2013, 42, 3689–3707 RSC
- K. Moeller, B. Yilmaz, R. M. Jacubinas, U. Mueller and T. Bein, J. Am. Chem. Soc., 2011, 133, 5284–5295 CrossRef PubMed
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC
- Z. K. Xie, Z. C. Liu, Y. D. Wang, Q. H. Yang, L. Y. Xu and W. P. Ding, Int. J. Mol. Sci., 2010, 11, 2152–2187 CrossRef CAS PubMed
- K. Na, M. Choi and R. Ryoo, Microporous Mesoporous Mater., 2013, 166, 3–19 CrossRef CAS PubMed
- M. Y. Kustova, P. Hasselriis and C. H. Christensen, Catal. Lett., 2004, 96, 205–211 CrossRef CAS
- H. Wang and T. J. Pinnavaia, Angew. Chem., Int. Ed., 2006, 45, 7603–7606 CrossRef CAS PubMed
- D. P. Serrano, T. J. Pinnavaia, J. Aguado, J. M. Escola, A. Peral and L. Villalba, Catal. Today, 2014, 227, 15–25 CrossRef CAS PubMed
- D. P. Serrano, R. A. Garcia, G. Vicente, M. Linares, D. Prochazkova and J. Cejka, J. Catal., 2011, 279, 366–380 CrossRef CAS PubMed
- K. Zhu, J. Sun, J. Liu, L. Wang, H. Wan, J. Hu, Y. Wang, C. H. F. Peden and Z. Nie, ACS Catal., 2011, 1, 682–690 CrossRef CAS
- Y. Zhang, K. Zhu, X. Zhou and W. Yuan, New J. Chem., 2014, 38, 5808–5816 RSC
- Z. P. Kong, B. Yue, W. Deng, K. K. Zhu, M. G. Yan, Y. F. Peng and H. Y. He, Appl. Organomet. Chem., 2014, 28, 239–243 CrossRef CAS
- J. Zhu, Y. Zhu, L. Zhu, M. Rigutto, A. van der Made, C. Yang, S. Pan, L. Wang, L. Zhu, Y. Jin, Q. Sun, Q. Wu, X. Meng, D. Zhang, Y. Han, J. Li, Y. Chu, A. Zheng, S. Qiu, X. Zheng and F. S. Xiao, J. Am. Chem. Soc., 2014, 136, 2503–2510 CrossRef CAS PubMed
- F. S. Xiao, L. F. Wang, C. Y. Yin, K. F. Lin, Y. Di, J. X. Li, R. R. Xu, D. S. Su, R. Schlogl, T. Yokoi and T. Tatsumi, Angew. Chem., Int. Ed., 2006, 45, 3090–3093 CrossRef CAS PubMed
- L. Xu, C. G. Li, K. Zhang and P. Wu, ACS Catal., 2014, 4, 2959–2968 CrossRef CAS
- J. C. Groen, J. A. Moulijn and J. P. Ramirez, Ind. Eng. Chem. Res., 2007, 46, 4193–4201 CrossRef CAS
- M. Milina, S. Mitchell, P. Crivelli, D. Cooke and J. P. Ramirez, Nat. Commun., 2014, 5, 1–10 Search PubMed
- M. S. Holm, E. Taarning, K. Egeblad and C. H. Christensen, Catal. Today, 2011, 168, 3–16 CrossRef CAS PubMed
- M. Milina, S. Mitchell, D. Cooke, P. Crivelli and J. P. Ramírez, Angew. Chem., Int. Ed., 2014, 5, 1591–1594 Search PubMed
- K. Li, J. Valla and J. G. Martinez, ChemCatChem, 2014, 6, 46–66 CrossRef CAS
- J. M. Thomas and R. K. Leary, Angew. Chem., Int. Ed., 2014, 53, 12020–12021 CrossRef CAS PubMed
- P. Kortunov, S. Vasenkov, J. Karger, R. Valiullin, P. Gottschalk, M. F. Elia, M. Perez, M. Stocker, B. Drescher, G. McElhiney, C. Berger, R. Glaser and J. Weitkamp, J. Am. Chem. Soc., 2005, 127, 13055–13059 CrossRef CAS PubMed
- C. A. Fyfe, H. Gies, G. T. Kokotailo, C. Pasztor, H. Strobl and D. E. Cox, J. Am. Chem. Soc., 1989, 111, 2470–2474 CrossRef CAS
- M. Y. Kustova, S. B. Rasmussen, A. L. Kustov and C. H. Christensen, Appl. Catal., B, 2006, 67, 60–67 CrossRef CAS PubMed
- H. Zhu, Z. Liu, D. Kong, Y. Wang and Z. Xie, J. Phys. Chem. C, 2008, 112, 17257–17264 CAS
- H. L. Chen, J. Ding and Y. M. Wang, New J. Chem., 2014, 38, 308–316 RSC
- A. Karlsson, M. Stöcker and R. Schmidt, Microporous Mesoporous Mater., 1999, 27, 181–192 CrossRef CAS
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodriguez and A. Peral, Chem. Mater., 2006, 18, 2462–2464 CrossRef CAS
- D. P. Serrano, R. Sanz, P. Pizarro and I. Moreno, Appl. Catal., A, 2012, 435, 32–42 CrossRef PubMed
- W. Xu, J. Dong, J. Li, J. Li and F. Wu, J. Chem. Soc., Chem. Commun., 1990, 10, 755–756 RSC
- M. Matsukata, N. Nishiyama and K. Ueyama, Microporous Mater., 1996, 7, 109–117 CrossRef CAS
- M. Matsukata, T. Osaki, M. Ogura and E. Kikuchi, Microporous Mesoporous Mater., 2002, 56, 1–10 CrossRef CAS
- M. H. Kim, H. X. Li and M. E. Davis, Microporous Mater., 1993, 1, 191–200 CrossRef CAS
- J. Chen, W. Hua, Y. Xiao, Q. Huo, K. Zhu and X. Zhou, Chem.–Eur. J., 2014, 20, 14744–14755 CrossRef CAS PubMed
- N. S. Nesterenko, F. Thibault-Starzyk, V. Montouilliout, V. V. Yushchenko, C. Fernandez, J. P. Gilson, F. Fajula and I. I. Ivanova, Kinet. Catal., 2006, 47, 40–48 CrossRef CAS
- C. Weidenthaler, Nanoscale, 2011, 3, 792–810 RSC
- J. C. Groen, L. A. A. Peffer and J. P. Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS
- F. T. Starzyk, I. Stan, S. Abello, A. Bonilla, K. Thomas, C. Fernandez, J. P. Gilson and J. P. Ramirez, J. Catal., 2009, 264, 11–14 CrossRef PubMed
- K. Moller, B. Yilmaz, U. Muller and T. Bein, Chem.–Eur. J., 2012, 18, 7671–7674 CrossRef PubMed
- H. Colfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef PubMed
- J. Fan, C. Z. Yu, T. Gao, J. Lei, B. Z. Tian, L. M. Wang, Q. Luo, B. Tu, W. Z. Zhou and D. Y. Zhao, Angew. Chem., Int. Ed., 2003, 42, 3146–3150 CrossRef CAS PubMed
- H. Cölfen and M. Antonietti, in Mesocrystals and Nonclassical Crystallization, ed. H. Cölfen and M. Antonietti, Wiley, Chichester, 2008, ch. 1, pp. 1–6 Search PubMed
- F. Wang, V. N. Richards, S. P. Shields and W. E. Buhro, Chem. Mater., 2013, 26, 5–21 CrossRef
- T. M. Davis, T. O. Drews, H. Ramanan, C. He, J. S. Dong, H. Schnablegger, M. A. Katsoulakis, E. Kokkoli, A. V. McCormick, R. L. Penn and M. Tsapatsis, Nat. Mater., 2006, 5, 400–408 CrossRef CAS PubMed
- G. Delahay, M. Mauvezin, B. Coq and S. Kieger, J. Catal., 2001, 202, 156–162 CrossRef CAS
- E. P. Parry, J. Catal., 1963, 2, 371–379 CrossRef CAS
- O. A. Anunziata, G. A. Eimer and L. B. Pierella, Appl. Catal., A, 2000, 190, 169–176 CrossRef CAS
- E. L. First, C. E. Gounaris, J. Wei and C. A. Floudas, Phys. Chem. Chem. Phys., 2011, 13, 17339–17358 RSC
- F. T. Starzyk, A. Vimont and J. P. Gilson, Catal. Today, 2001, 70, 227–241 CrossRef
- S. A. Khattaf and H. de Lasa, Appl. Catal., A, 2002, 226, 139–153 CrossRef
- P. M. Pacheco, J. M. Domínguez, L. Bucio, F. Alvarez, U. Sedran and M. Falco, Catal. Today, 2011, 166, 25–38 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2015 |