
Amine-functionalized magnetic nanocomposite particles for efficient immobilization of lipase: effects of functional molecule size on properties of the immobilized lipase†
Parvaneh Esmaeilnejad-Ahranjaniab,
Mohammad Kazemeinib,
Gurvinder Singhc and
Ayyoob Arpanaei*a
aDepartment of Industrial and Environmental Biotechnology, National Institute of Genetic Engineering and Biotechnology, PO Box: 14965/161, Tehran, Iran. E-mail: arpanaei@yahoo.com; Fax: +98-21-4458039; Tel: +98-21-44580463
bDepartment of Chemical and Petroleum Engineering, Sharif University of Technology, Tehran, Iran
cDepartment of Chemical Engineering, Norwegian University of Science and Technology, Trondheim, Norway
First published on 2nd April 2015
Abstract
A cost-effective design of reusable enzyme-functionalized particles with better catalytic activity is of great scientific interest due to their applications in a wide range of catalytic reactions in several industrial processes. In this work, a systematic approach for preparing amine-functionalized magnetic nanocomposite particles through the surface modification of core/shell type Fe3O4 cluster@SiO2 particles by the small molecules of 3-(2-aminoethyl)aminopropyltrimethoxysilane (AAS) or the large molecules of polyethyleneimine (PEI) with two different molecular weights, as the support materials for enzyme immobilization, has been demonstrated. The functional nanocomposite particles were characterized by STEM, XRD, EDX, VSM, TGA, FTIR, oxygen elemental analysis and zeta potential measurement techniques. Lipase from Pseudomonas cepacia, chosen as a model enzyme, was covalently immobilized on glutaraldehyde-activated particles. The free and immobilized lipases were characterized by UV-vis, FTIR and CD spectroscopic methods. It has been shown that the size of the functional molecule has a significant effect on the concentration of binding sites on the particles and consequently on the lipase immobilization efficiency and loading capacity as well as the conformation and activity of the immobilized lipase. Resulting from the increased binding site concentration on the low and high molecular weight PEI-functionalized particles' surface, high lipase immobilization efficiencies (87 and 97%, respectively) and loading capacities (803 and 817 mg g−1, respectively) were obtained. Upon the immobilization of lipase, the activity, thermal and storage stability as well as reusability were improved under harsh reaction conditions in the order of the lipase immobilized on the low molecular weight PEI > high molecular weight PEI > AAS functionalized particles. This study offers insight into the design of functionalized magnetic particles for efficient immobilization of enzymes as well as improvement of the immobilized enzymes properties.
Introduction
Enzymes are widely employed to catalyze a broad range of reactions in industry.1 However, their application in native form is often limited, mainly because of their short lifetime, and difficulties associated with their recovery and reusability making the processes less economically feasible.2,3 Immobilization of enzymes on various supports has been considered to overcome these problems by providing a more stable, recoverable and reusable biocatalyst from the reaction media.4,5 Nanoscale materials can potentially be used as supports for enzyme immobilization due to their size-dependent unique physical properties and high specific surface area.6 Among various materials, iron oxide nanoparticles have been greatly favored because of their easy and inexpensive synthesis, low toxicity and rapid magnetic separation from a reaction media averting the use of expensive liquid chromatography, filters, centrifuges and other instruments for the separation.7,8 In particular, magnetite nanoparticles with a size below 20 nm have attracted more attention due to their superparamagnetic behavior avoiding magnetically induced self-aggregation of particles.9,10 However, the iron oxide nanoparticles are chemically unstable under acidic and oxidative industrial reaction environments.9,11 Therefore, coating of a protective layer on the magnetic particles is necessary to maintain their stability.12,13 Design of composite materials such as a core/shell morphology encapsulating magnetic nanoparticles inside a shell of silica can be effective for extension of their applications due to the unique characteristics of silica shells including high stability under extreme reaction condition, good dispersion in aqueous solution, biocompatibility and ability to provide a platform for further surface modification due to abundant hydroxyl groups on the silica surface.7,14,15Moreover, different factors can influence the conformation and activity of the immobilized enzyme on nanoparticles, such as immobilization techniques, concentration of functional groups on the particles surface and reaction physico-chemical parameters.16,17 Among the different enzyme immobilization methods, covalent immobilization has been reported as a better approach to bind enzyme molecules onto the solid support providing no or negligible enzyme leakage.18,19 The number of functional groups on the surface of solid support can be an important parameter in terms of achieving the desired enzyme stability, conformation and activity under relevant physico-chemical reaction condition as well as the appropriate enzyme loading capacity and immobilization efficiency.17,20 Various methods have been explored for the chemical binding of enzymes to magnetic particles functionalized with small functional molecules such as aminopropyltriethoxy silane (APTES),21 3-glycidoxypropyltrimethoxysilane (GLYMO),22 3-glycidoxypropyltrimethoxysilane (GPTMS) and 3-mercaptopropyltrimethoxysilane (MPTMS).23 However, these methods usually result in low loading capacities of the particles for enzyme immobilization and poor conformational and catalytically stabilities of the immobilized enzymes.
An alternative solution can be functionalization of particles surface by polymer molecules which enhance enzyme immobilization efficiency and loading capacity of the particles through providing a high concentration of functional groups on the particles surface.18,24,25 It is also expected that the high number of covalent bonds between the polymer-functionalized particles surface and the enzyme molecules result in the immobilized enzymes with stable conformations and activities under harsh reaction conditions.26–29 Adsorption of the polymer molecules on the surface of magnetic particles via electrostatic interactions has been reported as a simple approach for surface modification;30 however, electrostatic interactions are not strong enough and vary by changes of temperature or pH resulting in desorption of the polymer molecules from the particles surface.31,32 Plasma polymerization has also been used as an alternative strategy for modifying the particle surface with polymer molecules.7,27,33 However, this process is a very complex technique and less efficient for industrial scale modification of submicron particles. Therefore, despite numerous previous studies for modification of the particle surface with polymers, there is still demand for a simple and cost-effective approach to obtain a core/shell type polymer-functionalized nanocomposite particles with a stable polymer layer. Moreover, the effects of parameters such as polymer molecular weight and the concentration of binding sites on the enzyme loading capacity of the polymer-functionalized particles as well as on the immobilized enzyme's conformation and activity-related properties are still needed to be elucidated.
Herein, we report on preparation and characterization of amine-functionalized magnetic nanocomposite particles with a core/shell structure for enzyme immobilization. In the first step of synthesis, oleic acid-coated magnetite (Fe3O4) nanoparticles were synthesized. Magnetite particles were then coated and stabilized with a nonporous silica layer. PEI molecules, with two different molecular weights, were anchored to the as-prepared magnetic silica composite (MS) particles via a simple covalent attachment approach. The particles modified by a small molecule of AAS were also prepared. To demonstrate the utility of these functionalized composite particles as magnetically separable supports for immobilization of enzymes, lipase, which has a wide range of industrial applications,1,34 was chosen as a model enzyme and covalently immobilized on the functionalized particles surface using glutaraldehyde (GA) as the cross-linker agent. The sequential steps involved in the preparation of nanocomposite particles and lipase immobilization are illustrated in Scheme 1a. The correlation between the particles functional parameters and properties of the immobilized enzyme is illustrated in Scheme 1b. We studied the effects of functional molecule size on the binding sites concentration of particles and consequently on the enzyme loading capacity and immobilization efficiency, as well as conformation, activity, stability, reusability and catalytic reaction kinetics of the immobilized lipase.
 | ||
| Scheme 1 Schematic illustration of (a) the synthesis of amine-functionalized magnetic silica nanocomposite particles and lipase immobilization; (b) the correlation between particles functional parameters and properties of the immobilized enzyme. AAS; 3-(2-aminoethylamino)propyltrimethoxysilane, GA; glutaraldehyde, PEI; polyethyleneimine. | ||
Experimental
Materials
Ferrous chloride tetrahydrate (FeCl2·4H2O), ferric chloride hexahydrate (FeCl3·6H2O), oleic acid (99 vol%), ammonia (25 vol%), ethanol (99.9 vol%), toluene (99.9 vol%), tetraethyl orthosilicate (TEOS, 99 vol%), 3-(2-aminoethyl amino)propyltrimethoxysilane (AAS, 97 vol%), glutaraldehyde (GA, 25 vol%) were purchased Merck. Polyethyleneimine (PEI1, average molecular weight 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 99 wt%) was received from Alfa-Aesar. 4-Nitrophenyl palmitate (pNPP), polyethyleneimine (PEI2, average molecular weight 750000, 50 wt% in H2O) and lipase from Pseudomonas cepacia were purchased from Sigma-Aldrich. All reagents were used as received without further purification. Milli-Q water (18.2 MΩ cm) was used throughout the work.
000, 99 wt%) was received from Alfa-Aesar. 4-Nitrophenyl palmitate (pNPP), polyethyleneimine (PEI2, average molecular weight 750000, 50 wt% in H2O) and lipase from Pseudomonas cepacia were purchased from Sigma-Aldrich. All reagents were used as received without further purification. Milli-Q water (18.2 MΩ cm) was used throughout the work.
Preparation of Fe3O4 nanoparticles
Oleic acid-coated magnetite nanoparticles were prepared by the co-precipitation method as described previously.4,35 Briefly, 540.0 mg of FeCl3·6H2O together with 199.0 mg of FeCl2·4H2O was added to a 150 mL three-necked flask and mixed with 60 mL of water under a nitrogen atmosphere with mechanical stirrer at 1000 rpm. Oleic acid (0.1 mL) and ammonia (7.5 mL) were added to the solution and the resulting mixture was stirred and heated to 80 °C. Then, 0.4 mL of oleic acid was continuously added to the solution in four steps every 5 min. The reaction was then allowed to proceed at 80 °C and 1000 rpm for 30 min. The resulting dark brown suspension was cooled down to room temperature and the nanoparticles were collected with a magnet and washed with water several times to remove nonmagnetic by-products.Preparation of Fe3O4 cluster@SiO2 (MS) nanocomposite particles
The synthesis of magnetite/silica core/shell nanocomposite (MS) was performed by the Stöber method via the hydrolysis-condensation of TEOS in the presence of Fe3O4 nanoparticles,36 with slight modifications. Briefly, the as-prepared magnetite nanoparticles (75 mg) were dispersed in toluene (5 mL) and then re-dispersed in a mixed solution of ethanol (80 mL), deionized water (20 mL) and ammonia (2.5 mL). Emulsion drops were formed by ethanol as the emulsifier once toluene suspended particles were added to the ethanol. The mixed solution was homogenized at room temperature and 400 rpm for 20 min to form a uniform dispersion of nanoparticles. Then, 1 mL of TEOS was added to the solution by dropwise addition in 30 min under stirring of 400 rpm at room temperature. The resulting mixture was stirred at room temperature for 4 h to complete the synthesis of MS nanoparticles. The product was separated using a magnet and washed several times with ethanol and water to remove nonmagnetic by-products. The collected nanoparticles were dried at 50 °C.Preparation of ASS- and PEI-functionalized Fe3O4 cluster@SiO2 nanocomposite particles
The surface of MS particles was modified by AAS molecules to render a platform for covalently attachment of either enzyme molecules or PEI molecules. For synthesis of the AAS-functionalized MS (AAS-MS) particles, 5 mg of the as-prepared MS particles were dispersed in 4.6 mL of ethanol. Then, water (0.2 mL), acetic acid (0.1 mL) and AAS (0.1 mL) were added to the mixture under continuous stirring.37 The reaction was allowed to proceed for 1 h and then the product was collected with a magnet, washed excessively with ethanol and water and finally dispersed in an aqueous sodium phosphate buffer solution (2.5 mL). Unless it is otherwise specified, 0.1 mol L−1 sodium phosphate buffer solution (pH 7.4) was used throughout in the present study.To prepare PEI-functionalized particles through covalent attachment method, the AAS-MS particles were activated with GA through the following procedure. 100 μL of GA was added to 2.5 mL of 2 mg mL−1 solution of AAS-MS particles and the resulting solution was continuously shaken at 60 oscillations min−1 and 4 °C for 60 min. After the reaction, the GA-activated AAS-MS particles (hereafter shown as GA-AAS-MS) were washed three times with the buffer solution, and then dispersed in 2.5 mL of buffer solution. GA is known as the cross-linker agent which forms an imine bond in the reaction with primary amine groups and leaves the other aldehyde group free for further conjugation of molecules containing primary amine groups.23,38 Then, 2.5 mL of either 0.2 mmol L−1 solution of PEI1 or 2.7 × 10−3 mmol L−1 solution of PEI2 in the buffer solution were added to the GA-AAS-MS particles solution and allowed to shake at 60 oscillations min−1 for 2 h. The final mass concentration of both polymer solutions were 2 g L−1. The resulting PEI-functionalized particles, hereafter referred as PEI1-MS and PEI2-MS, were washed three times with a buffer solution and finally dispersed in a buffer solution (2.5 mL).
Lipase immobilization on functionalized MS particles
For immobilization of lipase molecules on the functionalized particles, the primary amine functional groups on the AAS-MS, PEI1-MS and PEI2-MS particles surface were first activated with GA. 100 μL of GA was added to 2.5 mL of the as-prepared functionalized particles solution and the resulting mixture was shaken at 60 oscillations min−1 and 4 °C for 60 min. Once amine groups were activated, the samples were washed three times with a buffer solution and dispersed in 2.5 mL of the buffer solution. The GA-activated particles hereafter are referred as GA-AAS-MS, GA-PEI1-MS and GA-PEI2-MS. Then, 2.5 mL of lipase solution with various concentrations (2, 4, 6, and 8 mg mL−1) was rapidly added to all the GA-activated particles solutions. According to the literature,4 lipase from Pseudomonas cepacia preserves its maximum activity at pH 7.4, hence the immobilization procedure was carried out at this pH value. After overnight incubation at 4 °C, the lipase-immobilized GA-AAS-MS (L-AAS-MS), GA-PEI1-MS (L-PEI1-MS) and GA-PEI2-MS (L-PEI2-MS) samples were first washed three times with a sodium phosphate buffer solution (pH 7.4). Since the surface charges of the particles and the enzyme are opposite at pH 7.4, the enzyme molecules can physically adsorb onto the particles surface via various interactions such as electrostatic attractive forces. Therefore, the samples were washed with acetate buffer solutions of two pH values (0.1 mol L−1, pH 3 and 5, twice with each solution) and then with sodium phosphate buffer solutions of three pH values (0.1 mol L−1, pH 6, 8 and 9, twice with each solution) to ensure the removal of all the noncovalently-attached or loosely-bounded lipases. The washed samples were finally dispersed in a phosphate buffer solution (pH 7.4) and stored at 4 °C.Immobilization efficiency (i.e., mass ratio of the immobilized lipase to the initial lipase, wt%) and loading capacity (i.e., mass ratio of the immobilized lipase to the functionalized particle, mg g−1) of all prepared samples were estimated by Bradford assay using bovine serum albumin as a standard protein by measuring the content of lipase in the original lipase solution, supernatant and collected washing solutions.
Catalytic activity assays
Lipase activity study was performed according to the procedure reported in the literature,24 with a slight modification. Briefly, 450 μL of 1 mmol L−1 4-nitrophenyl palmitate (pNPP) solution was added to 50 μL of 2 mg mL−1 solution of either free lipase or immobilized lipase in a buffer solution. All the reaction systems contained a similar amount of lipase in either free or immobilized state. The resulting mixture was shaken at 110 rpm for an equilibrium reaction time of 15 min. Control experiments were also performed on the buffer solutions and particles without the immobilized lipase. As accurate measurement of enzyme activity requires complete conversion of 4-nitrophenol product into 4-nitrophenoxide; hence, NaOH was added to the reaction mixture, after the completion of reactions and recovering the particles using a magnet, and the pH value of the solution was adjusted to 10 to ensure more than 99% ionization of the pNP product. The molar concentration of the hydrolysis product, i.e., p-nitrophenol (pNP), which is equal to the molar concentration of 4-nitrophenoxide, was measured using the spectrophotometry method (T80 + UV/VIS Spectrophotometer, PG Company, Germany) at 405 nm.39 One unit of lipase activity was defined as the amount of lipase liberating 1 μmol of pNP per minute under the assay conditions and referred to as specific activity (U mg−1).The kinetic parameters of Michaelis–Menten equation for the free and immobilized lipases were determined by measuring the initial rates of the hydrolysis reaction through varying the substrate (pNPP) concentration,40 in a range of 0.05–5 mmol L−1 at 40 °C and 50 °C, at a constant pH 7.4.
To investigate the effect of temperature on hydrolytic activity of the free and immobilized lipases, the hydrolysis reactions was carried out at different temperatures, ranging from 30 °C to 60 °C, at a constant pH 7.4.
Thermal stability of the free and immobilized lipases was determined by evaluating the residual enzymatic activities after incubation in a phosphate buffer solution (pH 7.4) at two different temperatures of 40 °C and 50 °C for 1–10 h. The storage stability of free and immobilized lipases was determined by measuring the catalytic activities at 40 °C after storing in a buffer solution at 4 °C for a certain period of time.
Reusability of the immobilized lipases was measured through repeated uses in the hydrolysis reaction at 50 °C and pH 7.4. The immobilized lipases were magnetically recovered and washed excessively with a buffer solution prior to be used in the next reaction batch.
Characterization techniques
Scanning transmission electron microscopy (STEM) coupled with energy dispersive X-ray (EDX) spectroscopy was performed using a Hitachi S-5500 operating at 30 kV on the particles. Samples for STEM analysis were prepared by the evaporating of a dilute solution of particles onto 300 mesh carbon-coated copper grid. Scanning electron microscopy (SEM) images were obtained on a scanning electron Seron Technology AIS-2100 microscope. The average values of core size, shell thickness and overall diameter of the particles, and the standard deviation were calculated by counting over 100 particles using the ImageJ software.Bruker AXS D8 advanced diffractometer with Cu Kα radiation (λ = 1.5418 Å) was used for collecting X-ray diffraction (XRD) patterns. Vibrating sample magnetometer (VSM, Meghnatis Daghigh Kavir Co., Iran) operating at room temperature was employed to measure the magnetic properties of the particles. The temperature dependence of the magnetization of nanoparticles under zero-field-cooled (ZFC) and field-cooled (FC) (applied field of 100 Oe) conditions was studied using a superconducting quantum interference device (SQUID) magnetometer (MPMS, Quantum Design). Zeta (ζ) potential values and hydrodynamic diameters of the particles were measured using a Malvern Zetasizer Nano instrument (S90, UK) equipped with the dynamic light scattering (DLS) system. Fourier-transform infrared (FTIR) spectra were recorded from a Bruker Tensor 27 FTIR spectrophotometer with a resolution of 4 cm−1 using pellets made by 1 mg samples in 100 mg KBr. Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) using TG/DTA6300 instrument was performed on the dried particles at a heating rate of 10 °C min−1 under air atmosphere. Sample studied by TGA were also analysed by CHNS–O Elemental Analyzer instrument (Costech ECS 4010, USA) for quantitative determination of oxygen content of the particles.
The secondary conformational characteristics of the free and immobilized lipase molecules were studied by circular dichroism (CD) method using a J-715 spectropolarimeter (Jasco, Japan) over the wavelength rage of 190–260 nm on 0.2 mg mL−1 solution of either free lipase or immobilized lipase in a sodium phosphate buffer solution (0.1 mol L−1, pH 7.4) at a desired temperature. Blank spectra of the buffer solution with and without particles were obtained at identical conditions for background correction.
Data analysis
Analysis of variance (ANOVA) was used to determine whether the effects of the examined parameters are statistically significant (p < 0.05). The significance of differences was analyzed by the Duncan test as a post-hoc test. All samples were analyzed in triplicate and mean ± standard deviation values were calculated and reported. The TGA, XRD, EDX, VSM, FTIR, oxygen elemental analysis and hydrodynamic diameter and zeta potential measurements were assessed in duplicate. The CD spectroscopy analysis was performed in triplicate.Results and discussion
Characterization of nanocomposite particles
Fig. 1a–e presents the electron microscopy images of the synthesized particles. The Fe3O4 nanoparticles have a uniform diameter of 7.8 ± 0.9 nm. The MS particles, which were obtained via encapsulating of magnetite nanoparticles in a shell of silica, are near-spherical (210 ± 50 nm) and possess a core/shell structure with a 130 ± 30 nm core and a 40 ± 10 nm silica shell. The AAS-MS, PEI1-MS and PEI2-MS nanocomposite particles exhibit the same morphology as that of the MS particles with a mean diameter of 210 ± 50 nm.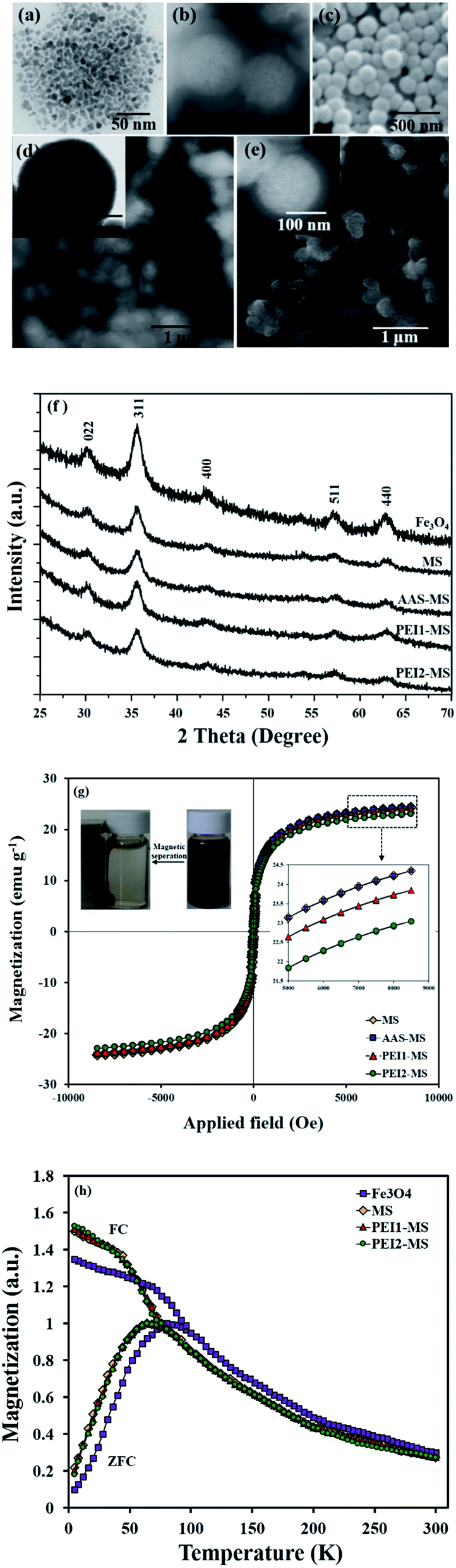 | ||
| Fig. 1 Electron microscopy images of magnetite (a), MS (b), AAS-MS (c), PEI1-MS (d) and PEI2-MS (e). XRD patterns (f), room temperature magnetization curves (g) and FC-ZFC curves (h) of bare and functionalized MS particles. | ||
The existence of 33 ± 4% Fe content in all the composite particles was verified from EDX spectra. Taking into account the mass (estimated from data obtained using TGA study, Fig. 2a and Table 1) and density of the organic/polymer layer, and the size of the MS particles, the thickness of organic layers on the dried PEI1-MS and PEI2-MS particles surface are calculated to be about 1.4 ± 0.2 nm and 2.3 ± 0.2 nm, respectively. The almost similar morphologies observed for all the samples indicates that the very thin organic/polymer layers were not detectable through imaging the particles using the electron microscopy. However, the size measurement through DLS revealed the larger diameters for PEI1-MS (310 ± 30 nm) and PEI2-MS (370 ± 30 nm) compared to the values determined from STEM images, most likely due to the swelling of the hydrophilic polymer layers in the aqueous solution. The hydrodynamic diameters of the MS (210 ± 30 nm) and AAS-MS (215 ± 30 nm) particles were almost similar to those determined from the electron microscopy images.
 | ||
| Fig. 2 TGA (a) and DTA (b) profiles of bare and functionalized MS particles. FTIR spectra (c) of pure PEI molecules, bare and functionalized MS particles. | ||
| Samples | AAS-MS | GA-AAS-MS | PEI1-MS | GA-PEI1-MS | PEI2-MS | GA-PEI2-MS |
|---|---|---|---|---|---|---|
| a Mass ratio of GA to MS particles calculated from oxygen elemental analysis results.b Mass ratio of GA to MS particles calculated from TGA analysis results. | ||||||
| Mass ratio of functional layer to MS particles (mF, mg g−1) | 31 | — | 87.12 | — | 188.12 | — |
| Concentration of functional molecules on particles (CF, μmol g−1) | 239 | — | 8.71 | — | 0.25 | — |
| Mass ratio of GA to MS particles (mGA, mg g−1) | — | 15.98a (17.68)b | — | 40.37 (46.03) | — | 88.32 (99.10) |
| Concentration of amine functional groups on particles (CNH2, μmol g−1) | 239 | — | 543 | — | 1366 | — |
| Concentration of binding sites on particles (CCHO, μmol g−1) | — | 190 | — | 480 | — | 1050 |
| Zeta (ζ) potential at pH 7.4 (mV) | −8.2 ± 1.3 | — | 15.7 ± 1.8 | — | 19.7 ± 2.0 | — |
Fig. 1f shows the XRD patterns of the as-prepared particles. The presence of six diffraction peaks in diffractogram of magnetite nanoparticles confirms the inverse spinel structure of Fe3O4 nanoparticles (JCPDS card no. 85-1436). These magnetite nanoparticles retain their crystal structure upon the encapsulation in the silica shell and the subsequent functionalization with organic/polymeric molecules. A small reduction in the intensity of the corresponding XRD peaks for the MS, AAS-MS, PEI1-MS and PEI2-MS particles can be attributed to the presence of an amorphous structure of silica shell, where it is seen that the very thin organic/polymeric layers do not more affect the corresponding peaks' intensity.
The room-temperature magnetization curves reveal the superparamagnetic behavior of all the as-prepared particles (Fig. 1g). The MS particles present a relatively high saturation magnetization (Ms) value (24.4 emu g−1), which supports the use of simple separation of the particles by an external magnetic field. The organic/polymeric layer on the MS particles have a negligible effect on the reduction of the Ms values for the AAS-MS, PEI1-MS and PEI2-MS because the corresponding layers thickness is negligible as compared to the silica shell thickness. The lower Ms value of the composite particles compared to the Fe3O4 nanoparticles, i.e., 60 emu g−1 reported in our previous work for the same magnetite particles,4 is due to the contribution of the nonmagnetic silica shell around the Fe3O4 particles. It should be mentioned that all the nanocomposite particles could be easily separated from the suspension of 5 mg mL−1 particles in water with a magnet within 30 s at room temperature (Fig. 1g inset) and then re-dispersed by shaking.
The superparamagnetic behavior of the as-prepared particles under the experimental conditions is also identified from FC-ZFC curves as the FC and ZFC curves coincide at high temperatures and exhibit a paramagnetic-like decay with the increase of the temperature (Fig. 1h). The FC and ZFC curves split with the decrease of the temperature; the ZFC curves reach maxima at 80 K and 65 K corresponding to the blocking temperatures of the magnetite and composite nanoparticles, respectively, and then start to decrease, while the FC curves keep increasing. All the bare and functionalized MS particles exhibit the similar FC and ZFC curves; hence, the shift in the blocking temperature for the composite particles to a lower value in comparison to that of the magnetite particles can be due to the rather thick nonmagnetic silica shell around the magnetite clusters. It has been reported that the nonmagnetic shell reduces the interactions established between the magnetic nanoparticles, altering the superparamagnetic anisotropy energy barrier, and causes a shift in the blocking temperature to lower values.41 The decreased magnetic interactions between the nanoparticles upon coating with silica is also evident in the slopes of ZFC curves below blocking temperature since the steeper slopes are observed for the composite particles as compared to the magnetite nanoparticles.
In order to estimate the amount of functional molecules on the surface of particles after each functionalization step, we applied TGA technique (Fig. 2a). DTA profiles are presented along with the TGA profiles to establish where the mass loss is occurring (Fig. 2b). For all the samples, the mass loss below 150 °C is mostly related to desorption of the water physically adsorbed to the surface as presented as the endothermic peak in DTA profiles. For the bare MS particles, no further mass loss or endothermic/exothermic peaks are observed, in agreement with the literature,42 revealing that the decomposition of silica does not occur at temperatures below 600 °C. For the AAS-MS and GA-AAS-MS particles, the mass losses occur between 150 °C and 300 °C, due to the thermal decomposition of the corresponding organic components, which are also evident as the exothermic peaks in the DTA profiles. TGA profiles of the as-prepared PEI-functionalized particles show mass losses between 200 °C and 500 °C, due to the thermal decomposition of PEI molecules and the organic component (i.e., AAS and GA) under the attached PEI molecules. The DTA profiles of the PEI-functionalized particles present wide exothermic peaks and do not show individual exothermic peaks for the combustion of polymeric and organic components separately. In agreement with results reported previously,13,31 the exothermic peak of PEI molecules is very wide. Therefore, these results imply that most likely the combustion of all organic layer components happens in a similar temperature range, hence, the small exothermic peaks related to the decomposition of AAS and GA molecules are hidden within the exothermic peak produced through the decomposition of PEI molecules. After activation of the PEI-functionalized particles with GA (GA-PEI1-MS and GA-PEI2-MS), the mass loss increases again due to the decomposition of the attached GA molecules. The DTA profiles of the GA-activated particles exhibit a sharp peak cantered at 200 °C associated with the decomposition of GA molecules and a wide shoulder corresponding to the thermal decomposition of other organic components.
The mass of the organic/polymeric layer on the MS particles after each functionalization step are summarized in Table 1. The obtained results reveal that the mass of the functional molecule on the MS particles surface (mF) increases as the size of the applied molecules enhances, i.e., PEI2-MS > PEI1-MS > AAS-MS.
However, the number of the attached functional molecules on the particles surface (CF, μmol g−1), calculated using eqn (1), changes in the converse order. This can be due to this fact that the steric/electrostatic repulsive interactions between organic molecules of higher molecular weights, i.e., PEI, are severe and prohibit these molecules to attach closely on the surface of particles, which leads to the lower concentration of attached molecules of high molecular weights (PEI) compared to the small molecules of AAS.
 | (1) |
It is well known that if a suitable concentration of GA is employed in the reaction of GA with surface primary amine groups, one of the aldehyde groups of GA forms an imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) bond and the other aldehyde group is left free, which can be employed in the formation of another imine bond with an amine group existing in protein or other types of molecules,23,38 as illustrated in Scheme 1a. However, due to homofunctional nature of the bis-aldehyde crosslinker (i.e., GA), the formation of bonds between both aldehyde groups with adjacent amine groups existing on the surface of particles is inevitable.23 Therefore, in order to estimate the exact concentration of the free aldehyde groups, which are called binding sites in this paper, present on the surface of the GA-activated particles, the oxygen elemental analysis were performed on the GA-AAS-MS, GA-PEI1-MS and GA-PEI2-MS. The oxygen content related to the free aldehyde groups on the aforementioned GA-activated particles was estimated after subtracting the oxygen content of the AAS-MS particles, taken as the reference material, (mO,ref) from that of the GA-activated particles (mO,GA). It is also mentioned that the oxygen elemental analysis results for the PEI1-MS and PEI2-MS particles represents no oxygen content once subtracting the oxygen content of the reference material (AAS-MS).
N) bond and the other aldehyde group is left free, which can be employed in the formation of another imine bond with an amine group existing in protein or other types of molecules,23,38 as illustrated in Scheme 1a. However, due to homofunctional nature of the bis-aldehyde crosslinker (i.e., GA), the formation of bonds between both aldehyde groups with adjacent amine groups existing on the surface of particles is inevitable.23 Therefore, in order to estimate the exact concentration of the free aldehyde groups, which are called binding sites in this paper, present on the surface of the GA-activated particles, the oxygen elemental analysis were performed on the GA-AAS-MS, GA-PEI1-MS and GA-PEI2-MS. The oxygen content related to the free aldehyde groups on the aforementioned GA-activated particles was estimated after subtracting the oxygen content of the AAS-MS particles, taken as the reference material, (mO,ref) from that of the GA-activated particles (mO,GA). It is also mentioned that the oxygen elemental analysis results for the PEI1-MS and PEI2-MS particles represents no oxygen content once subtracting the oxygen content of the reference material (AAS-MS).
The free aldehyde groups concentration (CCHO, mmol CHO g−1 MS particles), which is equal to the oxygen concentration, was estimated using eqn (2) and reported in Table 1.
 | (2) |
According to Table 1, the CCHO value increases as the size of the functional molecule enhances as the ratio of CCHO for GA-AAS-MS:GA-PEI1-MS:GA-PEI2-MS can be estimated to be 1:6.68:14.26. Although, as mentioned previously, CF decrease as the functional molecule size increase, the larger functional molecule possessing the more numbers of primary amine functional groups in one molecule, i.e., the ratio of primary amine groups on AAS:PEI1:PEI2 is 1:82:6198, provides a platform for binding of the more numbers of GA molecules and consequently leads to the higher value of CCHO.
To understand the possible crosslinking of the primary amine groups on the surface of the functionalized-MS particles through the activation with GA molecules, the concentration of free primary amine groups (CNH2, mmol NH2 g−1 MS particles) on the AAS-MS, PEI1-MS and PEI2-MS particles were estimated using eqn (3), as summarized in Table 1, and the obtained results were compared with the CCHO value for the corresponding GA-activated particles.
 | (3) |
Comparing the results obtained from eqn (2) and (3) indicates that the CCHO values for GA-AAS-MS, GA-PEI1-MS and GA-PEI2-MS are about 11–23% lower than the CNH2 values for AAS-MS, PEI1-MS and PEI2-MS (Table 1). These results can confirm the above hypothesis on the formation of some imine bonds between both aldehyde groups of GA molecules with primary amine groups present on the surface of AAS-MS, PEI1-MS or PEI2-MS particles.
To further verify this hypothesis, the mass of the attached GA molecules on the GA-activated particles (mGA (mg GA g−1 MS particles)) was calculated using the data obtained from the oxygen elemental analysis (eqn (4)), and then compared with those obtained from the TGA analysis.
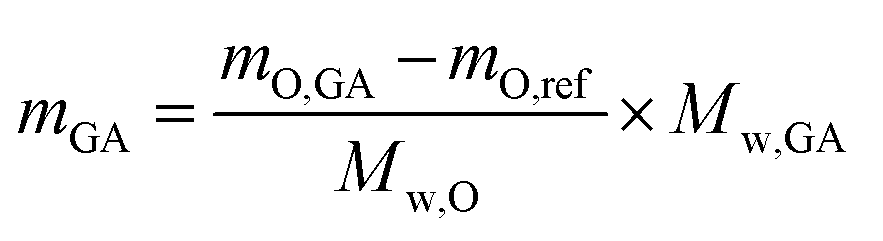 | (4) |
The results indicated that the mass of the attached GA molecules estimated from oxygen elemental analysis data (eqn (3)) is lower than that obtained using TGA analysis (Table 1), further confirming the above hypothesis. Because in the calculated mass of the GA on the particles from TGA analysis result, all GA molecules are taken into account regardless of having one free aldehyde group or both aldehyde groups taking part in the formation of imine bonds with surface amine groups. But, only the mass of the GA molecules with one free aldehyde group is measurable using the data obtained from oxygen elemental analysis method.
The functional groups of the modified and unmodified MS particles were further characterized by FTIR spectroscopy, as illustrated in Fig. 2c. For all the samples, the absorption band at 600 cm−1 is assigned to the vibration of Fe–O bonds.24 The stretching vibration of Si–O–Si, Si–OH and bending vibration of Si–O bonds make absorption peaks at 810, 973 and 1099 cm−1, respectively.26 The AAS-MS, PEI1-MS and PEI2-MS particles as well as the pure PEI1 and PEI2 molecules spectra exhibit two low-intensity absorption bands at 1617 and 1701 cm−1 associated with the bending vibration of N–H bonds and a peak at 3245 cm−1 corresponding to stretching vibration of N–H bonds in amine groups.8,43 The absorption peak at 1394 cm−1 is assigned to stretching vibration of C–N bonds.8 For the PEI1-MS and PEI2-MS, the stretching vibration of CN bonds make absorption peaks at 1488, 1570 and 1630 cm−1, verifying the covalent attachment of PEI molecules on the AAS-MS particles using GA as the cross-linker agent.43,44
The ζ potential values of the functionalized MS particles recorded at pH 7.4 are presented in Table 1. Alteration of surface charges of all the particles by changes in the pH of the solution is shown in Fig. S1.† The MS particles show a negative ζ potential value (−34.8 ± 2.3) due to the presence of a high number of hydroxyl groups on the surface in an aqueous medium.37 A considerable increase in ζ potential is noticed after modifying the particle surface with organic materials containing amine groups. The extent of increase in the ζ potential depends on the concentration of amine groups (CNH2, Table 1) introduced on the particles surface. The recorded ζ potential value for the AAS-MS particles is fairly in agreement with previous studies showing an almost similar change in ζ potential value of magnetic silica particles through functionalization with AAS, where the remained hydroxyl groups on the silica affects the surface charge of the amine-functionalized particles.7,37 The higher positive surface charge of the PEI2-MS particles as compared to the PEI1-MS ones can be attributed to high concentration of amine functional groups on the former.
Characterization of immobilized and free lipases
The conjugation of lipase onto the particles occurred via the formation of imine bonds between the aldehyde groups on the GA-activated particles and the amine groups in the lipase molecules as illustrated in Scheme 1a. After immobilization procedure, the samples were washed excessively with buffer solutions of various pH values to ensure the removal of the physically or electrostatically adsorbed lipase molecules. The total amount of unattached enzyme molecules collected after each washing step (Fig. S2†) was used to determine the lipase immobilization efficiency and loading capacity of the particles.The influence of lipase initial concentration on the immobilization efficiency and loading capacity are displayed in Fig. 3. A continuous decrease in the immobilization efficiency is found with the increase of lipase initial concentration, while the enzyme loading capacity initially increases with the increase of lipase concentration up to 2 mg mL−1 and then starts to decrease with further increase in the lipase initial concentration. As verified in the literature,14,45 strong protein–protein interactions lead to the formation of protein aggregates in the solution, limit the immobilization of lipases and consequently result in the reduction of the lipase loading capacity at the lipase concentrations above 2 mg mL−1, in this study. The results of lipase immobilization efficiency and loading capacity obtained at this lipase concentration (2 mg mL−1), which is used as the optimum value through this study, are summarized in Table 2. Obviously, there is a direct relation between the lipase immobilization efficiency or loading capacity and the binding site concentration on the particles at all the lipase initial concentrations. For instance, at 2 mg mL−1 enzyme initial concentration, the lipase immobilization efficiency and loading capacity ratios of L-AAS-MS:L-PEI1-MS:L-PEI2-MS particles are 1:1.77:1.97 and 1:1.68:1.72, respectively.
 | ||
| Fig. 3 Effect of lipase initial concentration on (a) lipase immobilization efficiency and (b) loading capacity of the amine-functionalized magnetic nanocomposite particles. | ||
| Samples | Free lipase | L-AAS-MS | L-PEI1-MS | L-PEI2-MS |
|---|---|---|---|---|
| a Mass percentage of the immobilized lipase to the initial lipase used for immobilization; the amount of immobilized lipase was calculated by subtracting the amount of un-immobilized lipase from the total amount of the lipase used for the immobilization.b Mass ratio of the immobilized lipase to the particles.c One unit of lipase activity defined as the amount of lipase liberating 1 μmol of pNP per minute under hydrolysis reaction of pNPP.d Specific activity per unit weight of lipase × enzyme loading capacity.e Kinetic parameters at reaction temperature of 40 °C.f Kinetic parameters at reaction temperature of 50 °C is shown in parentheses. | ||||
| Enzyme immobilization efficiencya (wt%) | — | 49.2 ± 0.4 | 87.3 ± 0.4 | 97.1 ± 0.3 |
| Enzyme loading capacityb (mg g−1) | — | 477 ± 4 | 803 ± 3 | 817 ± 2 |
| Molar ratio of binding sites on particles (CCHO) to immobilized lipase molecules (mol mol−1) | 11 | 21 | 36 | |
| Specific activity per unit weight of lipasec (U mg−1) | 2.32 ± 0.05 | 2.11 ± 0.03 | 1.98 ± 0.04 | 1.89 ± 0.03 |
| Specific activity ratio of immobilized lipase to free lipase (activity retention, %) | — | 91.3 ± 0.3 | 84.3 ± 0.2 | 78.8 ± 0.2 |
| Activity per unit weight of particled (U mg−1) | — | 1.0 ± 0.01 | 1.58 ± 0.03 | 1.54 ± 0.01 |
| Km (mmol L−1) | 0.53 ± 0.0e | 0.62 ± 0.05 | 0.83 ± 0.03 (0.44 ± 0.05)f | 1.07 ± 0.02 (0.78 ± 0.04) |
| Vmax (U mg−1) | 3.51 ± 0.0e | 3.29 ± 0.02 | 3.14 ± 0.05 (4.11 ± 0.03)f | 2.83 ± 0.03 (3.66 ± 0.04) |
The significantly higher values of lipase immobilization efficiency and loading capacity of the PEI1-MS and PEI-MS particles as compared to the AAS-MS particles can be attributed to higher binding sites concentration on the formers which create a microenvironment favoring more interactions and bond formations with the higher numbers of lipase molecules. The above mentioned ratios are not exactly according to the ratio of the binding sites concentration on the particles, i.e., 1:2.52:5.53 (Table 1), which could be due to steric barriers created by the large branched PEI molecules toward diffusion of the higher number of the lipase molecules within the polymer matrix. By calculating the number of lipase molecules on the surface of the AAS-MS particles (0.033 immobilized lipase molecules per nm2 AAS-MS) and taking into account the size of the lipase molecule (average diameter of 5 nm), it is revealed that the lipase molecules are packed closely together in one layer on the AAS-MS particles surface. In contrast, the high concentration of the lipase molecules on the PEI-functionalized particles should be dispersed within the binding sites of the large molecules of PEI. Moreover, taking into account the values of the binding site concentration and lipase loading capacity of the particles, the number of linking points for each lipase molecule conjugated onto the particles, represented as the molar ratio of the binding sites to the immobilized lipase molecules (Table 2), is estimated to be in the increasing order of L-PEI2-MS > L-PEI1-MS > L-AAS-MS.
FTIR spectra of the free and immobilized lipases are illustrated in Fig. 4a. The free lipase spectrum exhibits two absorption peaks at 1605 and 1707 cm−1 corresponding to C–O stretching vibration mode in amide I bonds and an absorption band at 1414 cm−1 associated with N–H bending mode and C–N stretching vibration mode in an amide II bonds. The bending of N–H bonds and stretching vibration of C–N bonds make absorption peak at 1323 cm−1 verifying the amide III bonds. The absorption peak at 2938 cm−1 is assigned to stretching vibration of C–H bonds.4,24 These bands are observed in the immobilized lipases spectra as well, indicating that the lipase was successfully attached on the surface of functionalized particles. For all the L-AAS-MS, L-PEI1-MS and L-PEI2-MS samples, the absorption bands at 1488 and 1570 cm−1 corresponding to CN vibration mode43,44 indicate the covalent attachment of lipase molecules to the particles using GA as the cross-linker agent.
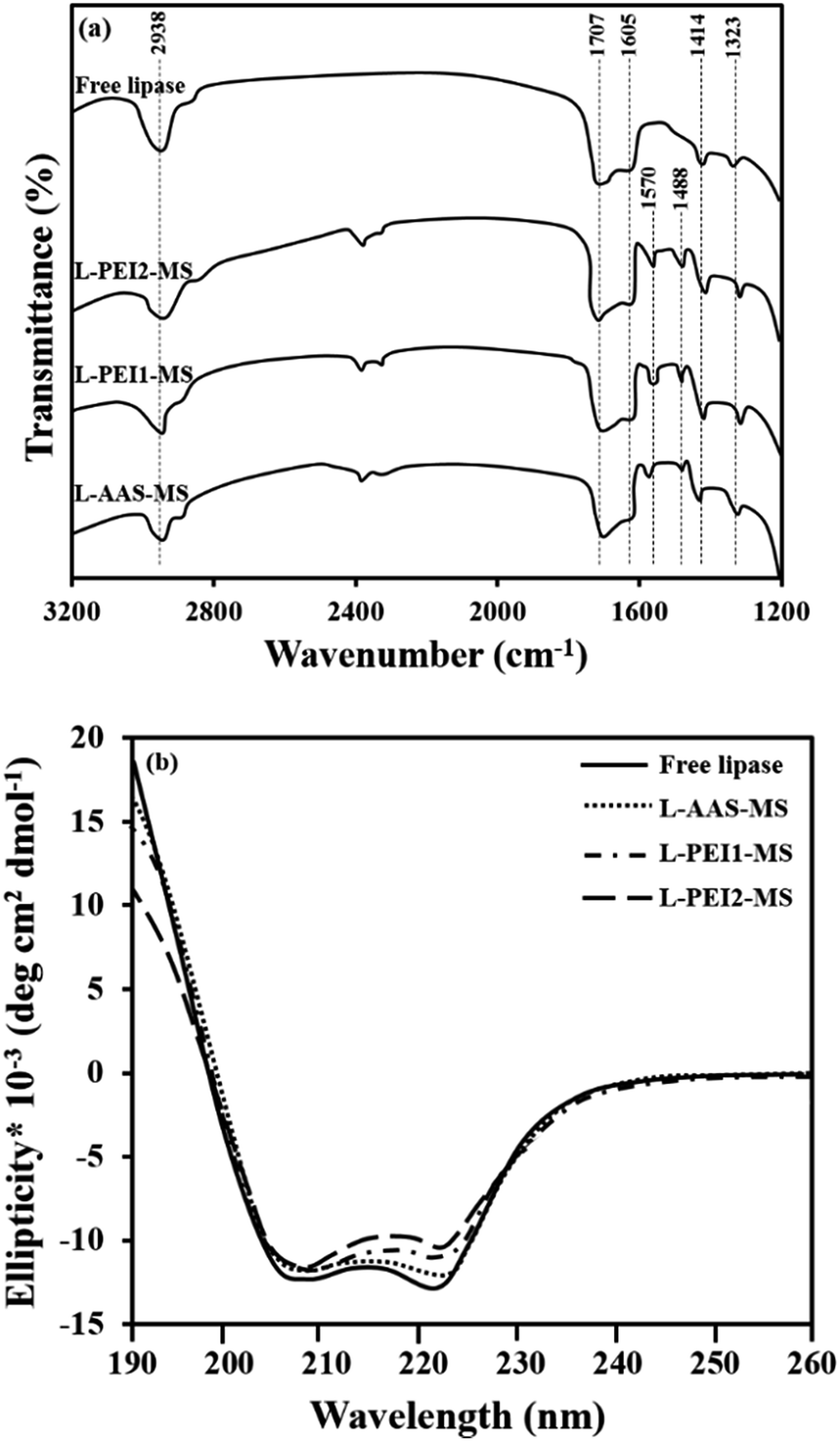 | ||
| Fig. 4 FTIR spectra (a) and CD spectra (b) of free and immobilized lipases. CD analysis condition: 2 mg mL−1 solution of fresh free or immobilized lipase in a sodium phosphate buffer solution (pH 7.4) stored at 4 °C. | ||
The CD spectroscopy was also performed on the free and immobilized lipases to provide information on the changes in the secondary conformation of the immobilized lipases as compared to the free lipase as a function of the available binding sites concentration on the functionalized particles surface. The CD spectra of the fresh free and immobilized lipases, suspended in buffer and stored at 4 °C for 15 min, exhibit a broad negative band from 208 to 222 nm (Fig. 4b), as the characteristic of enzyme molecules such as lipase, which contain both α-helices, making two intensive peaks at 208 and 222 nm, and β-sheets, making a peak at 217 nm.46,47 The proportional contents of secondary structural elements, i.e., α-helices, β-sheets, β-turns and random coils in the free and immobilized lipases are summarized in Table 3. Upon the immobilization of lipase, the α-helical and β-sheet contents partially decrease and the content of unordered elements marginally increases in the order of L-PEI2-MS > L-PEI1-MS > L-AAS-MS as compared to the free lipase. By considering the molar ratio of concentration of binding sites to the concentration of immobilized lipases (Table 2), it is revealed that there is a direct relation between the conformational changes of lipase and the number of covalent bonds between the lipase and particles. The alteration of enzyme conformation upon the covalent linkage onto the particles surface is well known, where the hydrogen bonds between the functional groups of an enzyme molecule, which give a specific structure for the enzyme molecule, are reduced as they are involved in the covalent linkage to the particles surface.20 Therefore, according to the literature,9 as the concentration of binding sites on the particles increases, the interactions between the lipase molecules and the particles increases and consequently the lipases suffers more conformational changes.
| Sample | α-Helix (%) | β-Sheet (%) | β-Turn (%) | Random coil (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a °C | 40b °C | 50c °C | R-50d °C | 4 °C | 40 °C | 50 °C | R-50 °C | 4 °C | 40 °C | 50 °C | R-50 °C | 4 °C | 40 °C | 50 °C | R-50 °C | |
| a Fresh lipases dispersed in a buffer solution at 4 °C and CD recorded at 4 °C.b Lipases pretreated in a buffer solution at 40 °C for 15 min and CD recorded at 40 °C.c Lipases pretreated in a buffer solution at 50 °C for 15 min and CD recorded at 50 °C.d Lipases recovered at the end of the 5th cycle of reusability assay performed at 50 °C and then dispersed in a buffer solution at 50 °C, and CD recorded at 50 °C. | ||||||||||||||||
| Free lipase | 31.6 | 31.4 | 22.7 | 20.4 | 20.3 | 16 | 18.1 | 18.3 | 25 | 29.9 | 30 | 33.3 | ||||
| L-AAS-MS | 30.2 | 29.4 | 23 | 18 | 19.9 | 19.1 | 15.5 | 10.6 | 18.9 | 20.5 | 24.5 | 30.9 | 31 | 31 | 35 | 40.5 |
| L-PEI1-MS | 28.1 | 27.9 | 21.5 | 21 | 18.7 | 19 | 25 | 23.3 | 20.5 | 21.6 | 20.5 | 22.6 | 32.7 | 32.5 | 32.6 | 33.1 |
| L-PEI2-MS | 26.3 | 24.5 | 17 | 15 | 17.1 | 17.7 | 23.1 | 19.1 | 23.4 | 24.8 | 25.9 | 29.2 | 33.2 | 35 | 34 | 36.7 |
The catalytic performance of the free and immobilized lipases was characterized at the optimum reaction condition (40 °C, pH 7.4, in this work) required for maximum activity of the free lipase from Pseudomonas cepacia, as common reaction conditions used to study the effect of immobilization on the catalytic properties of enzymes.27,48 The sodium phosphate buffer solution and the particles without immobilized lipase taken as controls showed no decomposition of substrate. The results summarized in Table 2 indicate that the specific activity per unit weight of lipase and specific activity retention follow the order of L-AAS-MS > L-PEI1-MS > L-PEI2-MS as compared to the free lipase. The immobilized lipases activities per unit weight of particles display a converse trend, indicating that as the lipase loading capacity of the particle increases, the lower amount of the corresponding particle is utilized to achieve a constant amount of lipase in the reaction media.
The variation of specific activity of lipase upon immobilization can be understood by looking into the reaction kinetic parameters. The kinetic characteristics of the free and immobilized lipases were also determined at this reaction condition (40 °C, pH 7.4), as illustrated in Fig. S3.†
The calculated Michaelis–Menten constant, i.e., Km, and the initial maximum reaction velocity, i.e., Vmax, values indicate that the affinity of enzymes for the substrate and the rate of reaction proceeding, decrease in the order of free lipase > L-AAS-MS > L-PEI1-MS > L-PEI2-MS (p < 0.05) (Table 2). Considering the CD analysis data recorded for the free and immobilized lipase at 40 °C which are not considerably different from the CD data recorded at 4 °C (Table 3), the lower affinity of immobilized lipases for substrate as compared to the free lipase is likely due to the conformational changes in the enzyme molecules upon immobilization onto the particles, which are in the order of L-PEI2-MS > L-PEI1-MS > L-AAS-MS. Conformational changes of enzyme molecules usually leads to the change in the active site and consequently the lower affinity of active site to the substrate molecules.20,27 Immobilization also led to the lower Vmax values for the immobilized lipases as compared to the free lipase. This phenomenon is commonplace in the enzyme immobilization process and is due to mass transfer limitation for substrate diffusion and accessibility to the active site of the immobilized enzymes.48,49 L-PEI1 and PEI2-MS exhibit the lower Vmax values as compared to L-AAS-MS because of the higher hampered accessibility of substrate molecules to the active sites of immobilized lipases within the large molecules of PEI. On the other hand, the lipases immobilized on the AAS-MS particles, which are scattered in one layer on the particles, are directly exposed to the substrate molecules. Such changes in the kinetic parameters of the lipase upon immobilization lead to the reduction of the specific activities of the immobilized lipases. However, the Vmax values for the immobilized lipases are about 80–93% of that for the free lipase, fairly implying that the resulting immobilized lipases can provide efficient catalytic ability similar to that of the free lipase.
Effect of temperature on activity of the free and immobilized lipases
Fig. 5 shows the effect of temperature on the hydrolytic activity of the free and immobilized lipases. The maximum activity value of the free and immobilized lipases at optimal conditions was defined as 100%, and the ratio of the specific activity to the maximum specific activity was referred as the relative activity. As seen in Fig. 5, the optimum temperature (i.e., the temperature at which the activity is maximum) of the tested samples varies from 40 °C to 55 °C in the order of L-PEI1-MS > L-PEI2-MS > L-AAS-MS > free lipase. From Fig. 5 inset, it is also observed that L-PEI1-MS and L-PEI2-MS exhibit 23 and 9% more activity at their own optimum temperatures (55 and 50 °C, respectively) than the native lipase at its optimum temperature (40 °C), while L-AAS-MS sample retains 95% of the native lipase activity at 45 °C as its optimum temperature. | ||
| Fig. 5 Effect of temperature on hydrolytic activity of free and immobilized lipases at pH 7.4. Inset: maximum activity of free and immobilized lipases at their own optimum temperatures; free lipase (40 °C), L-AAS-MS (45 °C), L-PEI1-MS (55 °C) and L-PEI2-MS (50 °C). | ||
The influence of temperature on the activity of enzymes is possibly raised from two distinct opposite phenomena. First, the hydrolytic reaction rate increases thermodynamically through increasing the temperature. Second, an increase in the temperature beyond the so-called optimum temperature leads to the changes in the enzyme conformation and reduces the enzyme activity negating the positive effect of temperature upon the reaction rate.8,27 The obtained results reveal that the conformational changes and the denaturation of the free lipase may occur at lower temperatures compared to the immobilized lipases.
The differences in the optimum temperatures and the effect of temperature on the lipases activities can be explained by considering the CD data of the free and immobilized lipases recorded after incubation in a buffer solution at 50 °C for 15 min (Table 3). The free lipase undergoes decreases of 8.4% and 4.3% in its α-helices and β-sheets, respectively, and equally an increase in the content of unordered structures at 50 °C as compared to those of the free lipase at 40 °C, suggesting the fast denaturation of the free lipase at temperatures above 40 °C as a main reason of its reduced activity.27,43
For L-AAS-MS, decreases of 6.4% and 3.6% in its α-helical and β-sheet contents are observed at 50 °C, respectively, as compared to those of L-AAS-MS at 40 °C, fairly implying the low conformational stability of L-AAS-MS at elevated temperatures. L-PEI1-MS does not show major changes in its unordered elemental contents and exhibits a decrease of 6% in its α-helical contents and equally an increase in the β-sheet contents at 50 °C as compared to those obtained at 40 °C. L-PEI2-MS shows a decrease of 5% in its α-helices and an increase of 5.3% in its β-sheets at 50 °C as compared to those obtained at 40 °C. Enhancement of the conformational stability of the covalently attached enzymes at elevated temperatures has also been documented in the literature.28,48,50 The more stable conformations of L-PEI1-MS and L-PEI2-MS compared to L-AAS-MS can also be related to rigidification of lipase molecules through multi-point covalent attachments with the large numbers of binding sites on the PEI1-MS and PEI2-MS particles (Table 1). The secondary structure of the lipases immobilized on PEI-functionalized particles was also studied at 55 °C. The L-PEI1-MS sample did not show significant changes in its conformation, but L-PEI2-MS represented decreases of 3% and 2.1% in the α-helical and β-sheet contents, respectively, as compared to those obtained at 50 °C. Owning to the conformational flexibility characteristic of the polymer chains,51 the more severe changes in the conformation of L-PEI2-MS compared to L-PEI1-MS may be due to the larger conformational changes of high molecular weight PEI molecules chains (PEI2) upon increasing the temperature compared to the low molecular weight PEI molecules (PEI1). The conformational changes of the PEI2 molecules may impose more striking conformational changes in the immobilized enzyme molecules and consequently lead to the lower activity of L-PEI2-MS at the reaction temperature of 55 °C as compared to L-PEI1-MS. However, further investigation is required to identify mechanisms governing the conformation changes in immobilized protein molecules imposed by conformational changes of polymer chains used as the immobilization matrix, upon increasing the temperature. These results also imply the importance of the β-sheet content and its crucial role in the retaining of enzyme activity upon immobilization at elevated temperatures. The L-PEI1-MS and L-PEI2-MS represent about 4.7% and 2.8% more β-sheets at 50 °C, respectively, as compared to the free lipase at 40 °C (Table 3). It can be concluded that the higher activities of L-PEI1-MS at 55 °C and L-PEI2-MS at 50 °C, compared to the free lipase at 40 °C, are resulted from not only the thermodynamically enhancement of the reaction rates at elevated temperatures, but also an increase in their β-sheet contents. According to the literature,46,52 the enhancement of β-sheet content in some enzymes leads to opening of the lipase's active site and increasing of the lipase affinity for substrate molecules. The kinetic parameters (Km and Vmax) of the lipases immobilized on PEI-functionalized particles also confirm the enhancement of these immobilized lipases affinity for substrate molecules and reaction rates at 50 °C (Table 2).
Thermal stability and storage ability of the free and immobilized lipases
Thermal stability of enzymes is one of the most important criteria for different applications.2 The inactivation courses of the free and immobilized lipases incubated in a buffer solution at 50 °C are shown in Fig. 6a. The relative activity is defined as the ratio of the measured specific activity to the initial specific activity. The free lipase is inactivated at a faster rate compared to the immobilized lipases as it preserves about 18% of its initial activity after 10 h heat treatment. L-AAS-MS, L-PEI1-MS and L-PEI2-MS retain 41, 81 and 72% of their initial activities, respectively. Thermal stability of free and immobilized lipases was also evaluated at 40 °C (Fig. S4†). All the free and immobilized lipases represent the rather more retained activities at 40 °C compared to those at 50 °C, i.e., 40, 76, 94 and 89% for free lipase, L-AAS-MS, L-PEI1-MS and L-PEI2-MS, respectively. However, as seen in Fig. 6a and S4,† the deactivation rate follows the same order of free lipase > L-AAS-MS > L-PEI2-MS > L-PEI1-MS at both temperatures. According to the literature,43 the higher thermal stability of the immobilized lipases compared to the free lipase can be due to the higher resistivity of the lipases against the denaturation through the covalent immobilization. The results of secondary structure of the free and immobilized lipases after incubation in a phosphate buffer solution at 50 °C for 10 h can confirm this hypothesis (Table S1†). For L-AAS-MS, L-PEI1-MS and L-PEI2-MS, decreases of 5.3%, 2.1% and 5% in the α-helical contents, and decreases of 8.1%, 2% and 4.2% in the β-sheet contents are observed after 10 h-heat treatment, respectively, as compared to those obtained after pre-treatment at 50 °C for 15 min. In contrast, the free lipase suffers more conformational changes and exhibits decreases of 15% and 9.5% in its α-helices and β-sheets, respectively, over the heat treatment as compared to those obtained after pre-treatment at 50 °C for 15 min. | ||
| Fig. 6 Thermal stability at 50 °C and pH 7.4 (a), and storage stability at pH 7.4 and 4 °C (b) for free and immobilized lipases. | ||
As seen in Fig. 6a and Table S1,† the higher thermal stabilities of the lipases immobilized on PEI-functionalized particles compared to L-AAS-MS can be due to the rigidification of their conformations through several covalent attachments on these particles (see molar ratio of binding sites to immobilized lipase molecules in Table 2). Enhanced thermal stability of enzymes through multi-point covalent bindings on particles has also been documented in the literature.23,53 The less thermal and conformational stability of L-PEI2-MS compared to L-PEI1-MS, is probably due to the changes in the conformation of the high molecular weight PEI molecules (PEI2) at elevated temperatures, as described in the previous section, which lead to the higher extent of conformational changes in the lipase molecules immobilized on the PEI2-MS particles.
Storage stability of the free and immobilized lipases was also examined through the measurement of the remaining lipase activity at 50 °C after storing in a buffer solution at 4 °C for 20 days (Fig. 6b). The free lipase and L-AAS-MS lose 60% and 47% of their initial activities within 20 days, respectively, while L-PEI1-MS and L-PEI2-MS retain almost 86 and 73% of their initial activities, respectively. The secondary structures of the free and immobilized lipase were also studied after storage in a phosphate buffer solution at 4 °C for 20 days (Table S1†). The obtained results indicate that the decreases in the α-helical and β-sheet contents are 1.4–9.3% and 1.6–6.8%, respectively, as compared to those estimated for the fresh ones, and follow the order of L-PEI1-MS < L-PEI2-MS < L-AAS-MS < free lipase. Compared to the free lipase and L-AAS-MS, the higher storage stabilities of the lipases immobilized onto the PEI-functionalized samples can be due to the multi-point covalent binding of the lipases on these particles, which retains the lipases in stable conformations.53
It should be mentioned that no detectable lipase molecules were observed in the immobilized lipases stocks prepared for the thermal and storage stability studies, as the Bradford assay was employed, verifying that the activity loss through these above-mentioned assays was not due to the possible leaching of the lipase molecules from the nanocomposite particles surface.
Reusability of the immobilized lipases
For development of a cost-effective enzymatic process, the reusability of immobilized lipases is an important issue.2 The reusability of the immobilized lipases through the hydrolysis reaction was easily performed by rapid separation and recovering the biocatalysts with a magnet. Fig. 7 demonstrates that L-AAS-MS, L-PEI1-MS and L-PEI2-MS have good reusability and retain nearly 57, 88 and 74% of their initial activities in the 5th cycle, respectively. In general, enzymes are very sensitive to environmental changes, and frequent treatment, i.e., recovering and washing with buffer and then using in a new reaction batch, would unavoidably make them denatured.13,27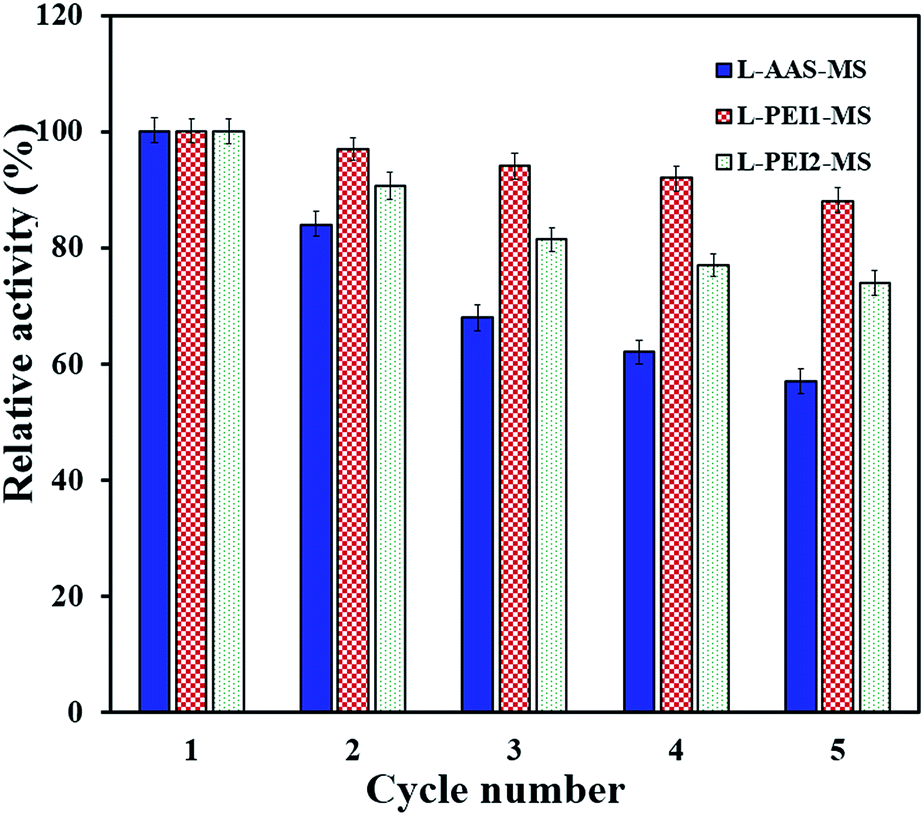 | ||
| Fig. 7 Reusability of the immobilized lipases at 50 °C and pH 7.4. | ||
To further investigate how the loss of activity can be related to the enzyme conformational changes, the secondary structure of the samples after 5th cycle was studied by CD analysis (Table 3). The changes in the conformation of the immobilized lipases, as compared to those pre-treated at 50 °C for 15 min (Table 3), are in the increasing order of L-AAS-MS > L-PEI2-MS > L-PEI1-MS. For L-AAS-MS, decreases of 5% and 4.9% in the α-helices and β-sheets can be observed, respectively, as compared to those obtained after pre-treatment at 50 °C for 15 min, revealing the unfolding of this lipase as a main reason of its activity loss over repeated uses. L-PEI1-MS suffers only decreases of 1% and 1.7% in its α-helices and β-sheets, respectively, while L-PEI2-MS undergoes decreases of 2% and 4% in its α-helices and β-sheets, respectively, after five times reusing as compared to those obtained after pre-treatment at 50 °C for 15 min. The formation of the larger number of multipoint covalent linkages between the PEI-functionalized particles surface and lipases leads to an increase in the conformational stability of lipases over a multi-cycle reuse as compared to the L-AAS-MS sample. As explained in the previous section, the lower stability of L-PEI2-MS compared to L-PEI1-MS over repeated uses may be related to the more severe conformational changes of the high molecular weight PEI molecules (PEI2) imposing higher extents of conformational changes in the enzyme molecules at a high temperature (50 °C, in this test).
Conclusions
In the present work, an easy, practical and low cost approach was employed to functionalize magnetic silica nanocomposite particles with the large molecules of PEI and the small molecules of AAS for lipase immobilization. We found that the size of the applied functional molecule is a crucial parameter strikingly influencing the properties of the functionalized particles and immobilized lipases. The binding site concentration, lipase immobilization efficiency, loading capacity and conformational change upon immobilization were directly correlated to each other and they varied in the order of L-PEI2-MS > L-PEI1-MS > L-AAS-MS. The activity-related assays revealed that the conformation, activity, thermal and storage stability as well as the reusability of the immobilized lipases were improved as compared to the free lipase, and increased in the order of L-PEI1-MS > L-PEI2-MS > L-AAS-MS.The enhanced activities and kinetic parameters of L-PEI1-MS and L-PEI2-MS at the elevated temperatures fairly suggested the direct correlation of the lipase activity and the β-sheet content, which increased in the order of L-PEI1-MS > L-PEI2-MS > L-AAS-MS as compared to the free lipase.
The obtained results demonstrated that an appropriate surface functionalization method is needed, such as PEI1-MS among the three samples presented in this work, to improve not only the loading capacity of the magnetic particles, but also the conformation and activity of the immobilized enzyme. This study opens up new perspectives for the design of polymer-modified nanoparticles for the efficient immobilization of enzymes with enhanced activity and stability.
Acknowledgements
This work is financially supported by the National Institute of Genetic Engineering and Biotechnology in Iran. The authors thankfully acknowledge the NTNU NanoLab for providing STEM characterization facility. The Research Council of Norway is acknowledged for the support to NTNU NanoLab through the Norwegian Micro- and Nano-Fabrication Facility, NorFab (197411/V30).Notes and references
- R. Diosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437 RSC.
- R. A. Sheldon and S. Pelt, Chem. Soc. Rev., 2013, 42, 6223 RSC.
- Y. Masuda, S. Kugimiya, Y. Kawachia and K. Kato, RSC Adv., 2014, 4, 3573 RSC.
- M. Kalantari, M. Kazemeini, F. Tabandeh and A. Arpanaei, J. Mater. Chem., 2012, 22, 8385 RSC.
- K. Murai, T. Nonoyama, T. Saito and K. Kato, Catal. Sci. Technol., 2012, 2, 310 Search PubMed; R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, Chem. Soc. Rev., 2013, 42, 6290 RSC.
- C. Garcia-Galan, A. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885 CrossRef CAS; E. T. Hwang and M. B. Gu, Eng. Life Sci., 2013, 13, 49 CrossRef; M. L. Verma, C. J. Barrow and M. Puri, Appl. Microbiol. Biotechnol., 2013, 97, 23 CrossRef PubMed.
- D. Shao, K. Xu, X. Song, J. Hu, W. Yang and C. Wang, J. Colloid Interface Sci., 2009, 336, 526 CrossRef CAS PubMed.
- Z. Chen, W. Xu, L. Jin, J. Zha, T. Tao, Y. Lina and Z. Wang, J. Mater. Chem. A, 2014, 2, 18339 CAS.
- H. H. P. Yiu and M. A. Keane, J. Chem. Technol. Biotechnol., 2012, 87, 583 CrossRef CAS.
- C. C. Berry and A. S. G. Curtis, J. Phys. D: Appl. Phys., 2003, 36, 198 CrossRef.
- Y. Saylan, L. Uzun and A. Denizli, Ind. Eng. Chem. Res., 2015, 54, 454 CrossRef CAS.
- X. Y. Liu, S. W. Zheng, R. Y. Hong, Y. Q. Wang and W. G. Feng, Colloids Surf., A, 2014, 443, 425 CrossRef CAS PubMed.
- Y. Yong, Y. Bai, Y. Li, L. Lin, Y. Cui and C. Xia, J. Magn. Magn. Mater., 2008, 320, 2350 CrossRef CAS PubMed.
- Y. Jiang, C. Guo, H. Xia, M. Iram, C. Liu and H. Liu, J. Mol. Catal. B: Enzym., 2009, 58, 103 CrossRef CAS PubMed.
- S. Abramson, C. Meiller, P. Beaunier, V. Dupuis, L. Perrigaud, A. Bee and V. Cabuil, J. Mater. Chem., 2010, 20, 4916 RSC.
- B. Saha, J. Saiki and G. Das, Analyst, 2015, 140, 532 RSC.
- O. Barbosa, R. Torres, C. Ortiz, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Biomacromolecules, 2013, 14, 2433 CrossRef CAS PubMed.
- L. Zang, J. Qiu, X. Wu, W. Zhang, E. Sakai and Y. Wei, Ind. Eng. Chem. Res., 2014, 53, 3448 CrossRef CAS.
- G. Zhang, J. Ma, J. Wang, Y. Li, G. Zhang, F. Zhang and X. Fan, Ind. Eng. Chem. Res., 2014, 53, 19878 CrossRef CAS.
- F. Secundo, Chem. Soc. Rev., 2013, 42, 6250 RSC.
- I. Koh, X. Wang, B. Varughese, L. Isaacs, S. H. Ehrman and D. S. English, J. Phys. Chem. B, 2006, 110, 1553 CrossRef CAS PubMed; Y. Li, X. Q. Xu, C. H. Deng, P. Y. Yang and X. M. Zhang, J. Proteome Res., 2007, 6, 3849 CrossRef PubMed; S. Lin, D. Yun, D. W. Qi, C. H. Deng, Y. Li and X. M. Zhang, J. Proteome Res., 2008, 7, 1297 CrossRef PubMed.
- S. Lin, Z. X. Lin, G. P. Yao, C. H. Deng, P. Y. Yang and X. M. Zhang, Rapid Commun. Mass Spectrom., 2007, 21, 3910 CrossRef CAS PubMed.
- D. Li, W. Y. Teoh, J. J. Gooding, C. Selomulya and R. Amal, Adv. Funct. Mater., 2010, 20, 1767 CrossRef CAS.
- J. Liu, W. Wang, H. Liu, Y. Zhou, H. Zhang and X. Zhou, RSC Adv., 2014, 4, 25983 RSC.
- Z. Qu, F. Hu, K. Chen, Z. Duan, H. Gu and H. Xu, J. Colloid Interface Sci., 2013, 398, 82 CrossRef CAS PubMed; X. Liu, X. Chen, Y. Li, X. Wang, X. Peng and W. Zhu, ACS Appl. Mater. Interfaces, 2012, 4, 5169 Search PubMed.
- Y. H. Lien and T. M. Wu, J. Colloid Interface Sci., 2008, 326, 517 CrossRef CAS PubMed.
- Y. Shen, W. Guo, L. Qi, J. Qiao, F. Wang and L. Mao, J. Mater. Chem. B, 2013, 1, 2260 RSC.
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262 RSC.
- Y. Ren, J. G. Rivera, L. He, H. Kulkarni, D. K. Lee and P. B. Messersmith, BMC Biotechnol., 2011, 11, 63 CrossRef CAS PubMed.
- K. Solanki and M. N. Gupta, New J. Chem., 2011, 35, 2551 RSC.
- H. H. P. Yiu, S. C. McBain, A. J. E. Haj and J. Dobson, Nanotechnology, 2007, 18, 435601 CrossRef.
- S. Chibowski, J. Patkowski and E. Grzadka, J. Colloid Interface Sci., 2009, 329, 1 CrossRef CAS PubMed.
- L. Lei, Y. X. Bai, Y. F. Li, L. X. Yi, Y. Yang and C. G. Xia, J. Magn. Magn. Mater., 2009, 321, 252 CrossRef CAS PubMed.
- P. Adlercreutz, Chem. Soc. Rev., 2013, 42, 6406 RSC.
- K. Tanaka, A. Narita, N. Kitamura, W. Uchiyama, M. Morita, T. Inubushi and Y. Chujo, Langmuir, 2010, 26, 11759 CrossRef CAS PubMed.
- W. Stober, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62 CrossRef.
- H. Mohammad-Beigi, S. Yaghmaei, R. Roostaazad, H. Bardania and A. Arpanaei, Phys. E, 2011, 44, 618 CrossRef CAS PubMed.
- G. Yang, J. Wu, G. Xu and L. Yang, Colloids Surf., B, 2010, 78, 351 CrossRef CAS PubMed.
- X. Wang, P. Dou, P. Zhao, C. Zhao, Y. Ding and P. Xu, ChemSusChem, 2009, 2, 947 CrossRef CAS PubMed.
- K. C. Badgujar and B. M. Bhanage, J. Phys. Chem., 2014, 118, 14808 CAS.
- A. B. Davila-Ibanez, N. J. Buurma and V. Salgueirino, Nanoscale, 2013, 5, 4797 RSC; A. B. Davila-Ibanez, V. Salgueirino, V. Martinez-Zorzano, R. Marino-Fernandez, A. Garcia-Lorenzo, M. Maceira-Campos, M. Munoz-Ubeda, E. Junquera, E. Aicart, J. Rivas, F. J. Rodriguez-Berrocal and J. L. Legido, ACS Nano, 2012, 6, 747 CrossRef CAS PubMed.
- Y. Huang, T. Hou, X. Cao, S. Perrier and Y. Zhao, Polym. Chem., 2010, 1, 1615 RSC; L. T. Zhuravlev, Colloids Surf., A, 2000, 173, 1 CrossRef CAS.
- B. Gao, X. Wang and Y. Shen, Biochem. Eng. J., 2006, 28, 140 CrossRef CAS PubMed.
- K. M. de Lathouder, D. T. J. van Benthem, S. A. Wallin, C. Mateo, R. Fernandez Lafuente, J. M. Guisan, F. Kapteijn and J. A. Moulijn, J. Mol. Catal. B: Enzym., 2008, 50, 20 CrossRef CAS PubMed.
- E. Ranjbakhsh, A. K. Bordbara, M. Abbasi, A. R. Khosropour and E. Shams, Chem. Eng. J., 2012, 179, 272 CrossRef CAS PubMed.
- T. Liu, Y. Liu, X. Wang, Q. Li, J. Wang and Y. Yan, J. Mol. Catal. B: Enzym., 2011, 71, 45 CrossRef CAS PubMed.
- A. Azizia, B. Ranjbar, K. Khajeh, T. Ghodselahic, S. Hoornamc, H. Mobasherid and M. R. Ganjalikhany, Int. J. Biol. Macromol., 2011, 49, 652 CrossRef PubMed.
- N. Ozturk, S. Akgol, M. Arisoy and A. Denizli, Sep. Purif. Technol., 2007, 58, 83 CrossRef CAS PubMed.
- Y. X. Bai, Y. F. Li, Y. Yang and L. X. Yi, Process Biochem., 2006, 41, 770 CrossRef CAS PubMed.
- P. Villeneuve, J. M. Muderhwa, J. Graille and M. J. Haas, J. Mol. Catal. B: Enzym., 2000, 9, 113 CrossRef CAS.
- J. Suh, S. H. Lee, S. M. Kim and S. S. Hah, Bioorg. Chem., 1997, 25, 221 CrossRef CAS; K. Holmberg, F. Tiberg, M. Malmsten and C. Brink, Colloids Surf., A, 1997, 123, 297 CrossRef.
- Y. Liu, X. A. Zhang, H. Tan, Y. J. Yan and B. H. Hameed, Process Biochem., 2010, 45, 1176 CrossRef CAS PubMed.
- J. Guo, W. Yang, C. Wang, J. He and J. Chen, Chem. Mater., 2006, 18, 5554 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Zeta potential values of the particles vs. pH; effect of the pH value of the buffer solution on removing the unattached lipase molecules; CD data of the free and immobilized lipases pre-treated according to the thermal and storage assays; hydrolysis reactions' kinetic curves; thermal stability of the lipases at 40 °C. See DOI: 10.1039/c5ra02471d |
| This journal is © The Royal Society of Chemistry 2015 |