DOI:
10.1039/C5RA02393A
(Paper)
RSC Adv., 2015,
5, 20656-20662
Green mechanical activation-assisted solid phase synthesis of cellulose esters using a co-reactant: effect of chain length of fatty acids on reaction efficiency and structure properties of products†
Received
7th February 2015
, Accepted 17th February 2015
First published on 17th February 2015
Abstract
Esterification is an important chemical modification for the preparation of natural cellulose-based materials. However, esterification of cellulose is commonly carried out in organic solvents, which reduces the economic and environmental feasibility of the synthetic technologies. Herein we report a novel technology for the production of cellulose esters, which combines mechanical activation (MA) and esterification in a stirring ball mill under solid phase conditions without the use of solvents. With the use of acetic anhydride as co-reactant and fatty acids as long chain esterifying agents, 1H and 13C NMR measurements confirmed that both acetyl and long chain fatty acyl groups were successfully grafted on cellulose by the technology of MA-assisted solid phase synthesis (MASPS). The factors which contributed to the successful preparation of cellulose esters were: the formation of highly reactive mixed acetic-long chain fatty acid anhydride, the generation of active hydroxyl groups in cellulose, the weakening of the steric effect of long chain fatty acids, and the improved contact between reagents and cellulose which were first induced by intense milling. It also showed that the reactivity of fatty acids and the degree of substitution (DS) of fatty acyls decreased with the increase in their chain length, and the long chain fatty acylium ions preferred to react with the more reactive hydroxyl group in the anhydro glucose unit of cellulose. Moreover, Fourier transform infrared spectroscopy, X-ray diffractometry, and scanning electron microscopy analyses were used to measure the changes in chemical structure, crystal structure, and surface morphology of the cellulose before and after esterification by MASPS, respectively. The results indicate that this green, simple, and efficient technology is suitable for the direct production of cellulose esters with long chain substituents.
1. Introduction
Natural cellulose is the most ubiquitous organic polymer in nature, and it is considered as an almost inexhaustible source of raw material for the preparation of renewable and biodegradable products.1 With the increasing consciousness of environmental protection and the dwindling supply of petroleum-based products, natural cellulose-based materials are being actively pursued.2,3 However, cellulose has limitations in its processability. It is neither meltable nor soluble in most common solvents because of the strong inter- and intramolecular hydrogen bonding and its high degree of crystallinity in structure.4–6 The properties and processability of cellulose can be dramatically modified by the substitution reactions of its hydroxyl groups, and cellulose is the most commercially significant nature polymer with billions of kilograms of its sales each year for a wide range of applications.7,8 Cellulose esters, which represent a class of commercially important thermoplastic polymers, are typical cellulose derivatives with excellent fiber and filmogenic characteristics.9 Moreover, cellulose esters can offer interesting alternatives to petrochemical plastics, depending on their physical properties.10 Unlike the case of other biopolymers, long chain cellulose esters (LCCE, chain length of fatty substituents ≥ C6) exhibit plasticized polymer behavior without any addition of external plasticizer.11 In order to eliminate fugitive plasticizers, the preparation of LCCE has afforded promising results.12 In general, esterification of cellulose is carried out in organic solvents, such as N,N-dimethylacetamide (DMAc)/LiCl, N,N-dimethylformamide (DMF)/LiCl, toluene/triethylamine (TEA), dimethyl sulfoxide (DMSO)/tetrabutylammonium fluoride trihydrate (TBAF), etc.4,13–15 The large amounts of used organic solvents are difficult to be effectively recovered, which reduces the economic feasibility of synthetic technologies and has harmful effects to the environment. Therefore, it is very important to develop a green, simple, and efficient method for the preparation of cellulose esters.
Solid phase synthesis (SPS) has been considered as an efficient procedure for green chemistry due to the advantages of nonuse of solvent, specificity of reaction, high selectivity and efficiency, saving resources, environmental friendliness, and simplicity of the technique.16–18 Accordingly, it is possible and promising to produce cellulose esters by SPS. However, cellulose possesses highly-ordered and stable crystalline structure ascribed to a network of inter- and intramolecular hydrogen bonds and van der Waals forces, which make it resists assault of other reagents, especially in SPS conditions.19 Generally, cellulose fatty acid esters are prepared with fatty acid chlorides as esterifying agents, but these powerful and relatively noxious reagents produce aggressive hydrochloric acid during esterification reaction. To limit the acid degradation of cellulose, pyridine and triethylamine are used to neutralize HCl as it is formed, or a nitrogen stream or vacuum is applied to remove gaseous HCl from the reaction system.20 The use of fatty acids as esterifying agents can avoid using these noxious reagents and complex methods, but the reactivity of fatty acids is very low, especially the long chain fatty acids which exhibit a steric effect when react with cellulose. Consequently, it is necessary to apply assisted means for enhancing the reactivity of cellulose and long chain fatty acids and the reaction efficiency of SPS.
Mechanical activation (MA), usually carried out by high-energy milling, refers to the use of mechanical actions to change the crystalline structure and physicochemical properties of the solids. MA is considered as a simple, efficient and environmentally friendly method for the pretreatment of solid materials, attributing to the use of simple and cheap equipment and the operations without the use of solvents, intermediate fusion, etc.21–23 When subjected to intense mechanical milling, the stable hydrogen bond and crystalline structure of cellulose were significantly destroyed, resulting in the increase of its reactivity.24 Moreover, during the process of MA, a part of mechanical energy can be converted into internal energy of the milled solids and thus generates many metastable active sites, which may rapidly reduce after removing the solids from the equipment of MA.25 In order to make full use of the mechanical energy induced by MA and improve the conditions of chemical modification by SPS, we combine MA and esterification of cellulose in the same equipment. This MA-assisted SPS (MASPS) method can simultaneously enhance the reactivity of cellulose and the contact between cellulose and reagents, leading to the simplification of the whole technological process and the increase of production efficiency. On the other hand, the reactivity of long chain fatty acids also should be improved. It has been reported that certain molecules, such as dicyclohexylcarbodiimide and 4-pyrrolidinopyridine, p-toluenesulfonyl chloride, methanesulfonyl chloride, trifluoroacetic anhydride, acetic anhydride, hydroquinone, or potassium acetate, are adopted to transform fatty acids into more reactive entities which are easy to graft on cellulose, and they can be used as co-reactant for enhancing the esterification of cellulose.20 Undoubtedly, the use of co-reactant can effectively improve the reactivity of long chain fatty acids. Therefore, the simultaneous assistance of MA and co-reactant can significantly enhance the SPS of LCCE.
In this study, MASPS technology was adopted for the esterification of cellulose with acetic anhydride as non-toxic co-reactant, long chain fatty acids as esterifying agents, and stirring ball mill as reactor. The cellulose mixed esters with acetyl and long chain fatty acyl groups could be directly produced by this simple and environmentally friendly method. The effect of chain length of fatty acids on reaction efficiency and properties of cellulose esters were detailedly investigated. To our knowledge, this is the first study on preparing cellulose mixed esters by MASPS with the use of co-reactant.
2. Experimental
2.1. Materials
Microcrystalline cellulose (MCC) with a degree of polymerization (DP) of 220 was obtained from Shanghai Yuanye Bio-Technology Co., Ltd. (China). Octanoic acid, lauric acid, palmitic acid, acetic anhydride, and other chemical reagents were of analytical grade without further purification and obtained commercially. Deionized water was used throughout the work.
2.2. Preparation of cellulose esters
The esterification of cellulose was performed in a customized stirring ball mill driven by a commercially available drill press equipped with a speed-tuned motor.26 In a typical experiment, a mixture of fatty acid, acetic anhydride, and H2SO4 catalyst (molar ratio of anhydro glucose unit (AGU) of cellulose![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fatty acid:acetic anhydride:H2SO4 = 1:3:6:0.06) was stirred for 60 min at 80 °C, and then 10.0 g of MCC was added and uniformly mixed. The resulting mixture was poured into a jacketed stainless steel chamber (1200 mL) which had been added a fixed amount of milling balls (400 mL, 5 mm diameter). The mixture was subjected to milling and reacted at the speed of 375 rpm under a constant temperature of 80 °C by circulating the thermostatic water in the jacket of chamber. When the mixture was milled for different designated time, the balls were removed from the resulting sample, which was then purified by a repeated washing-filtration process with water and absolute alcohol respectively. After oven-dried at 75 °C, the gray-white cellulose esters were obtained. Cellulose acetate–octanoate, cellulose acetate–laurate, and cellulose acetate–palmitate with different degree of substitution (DS) were prepared by this simple procedure by changing the reaction conditions.
:fatty acid:acetic anhydride:H2SO4 = 1:3:6:0.06) was stirred for 60 min at 80 °C, and then 10.0 g of MCC was added and uniformly mixed. The resulting mixture was poured into a jacketed stainless steel chamber (1200 mL) which had been added a fixed amount of milling balls (400 mL, 5 mm diameter). The mixture was subjected to milling and reacted at the speed of 375 rpm under a constant temperature of 80 °C by circulating the thermostatic water in the jacket of chamber. When the mixture was milled for different designated time, the balls were removed from the resulting sample, which was then purified by a repeated washing-filtration process with water and absolute alcohol respectively. After oven-dried at 75 °C, the gray-white cellulose esters were obtained. Cellulose acetate–octanoate, cellulose acetate–laurate, and cellulose acetate–palmitate with different degree of substitution (DS) were prepared by this simple procedure by changing the reaction conditions.
2.3. NMR analysis
1H and 13C NMR spectra were recorded on a Bruker AVANCE III 600 MHz spectrometer (Switzerland) at room temperature. For all NMR experiments, deuterated chloroform (CDCl3) and tetramethylsilane (TMS) were used as the solvent and internal standard, respectively. All chemical shifts were reported in parts per million (ppm). DS values of the cellulose esters were calculated by signal integration and correlation of cellulose and substituent protons in 1H NMR spectra of the products.27
2.4. Fourier transform infrared spectroscopy (FTIR) analysis
FTIR spectra of the samples were acquired on an 8400S Fourier transform infrared spectrometer (Shimadzu, Japan) using the KBr disk technique. For FTIR measurement, the samples were mixed with anhydrous KBr and then compressed into thin disk-shaped pellets. The spectra were obtained with a resolution of 4 cm−1 and a wavenumber range of 4000–400 cm−1.
2.5. X-ray diffractometry (XRD) analysis
XRD analysis was carried out by a UItima IV diffractometer (Rigaku, Japan) with Ni-filtered Cu Kα radiation (λ = 0.154 nm) at 40 kV and 40 mA. The measurements were conducted on powder compacted to small mats. XRD patterns were recorded from 5° to 35° with a step size of 0.02°.
2.6. Morphology observation
The changes in morphologies of the cellulose before and after MASPS were observed by scanning electron microscopy (SEM) analysis using an S-3400N scanning electron microscope (Hitachi, Japan). The samples were fixed on a sample bench using double-sided tape, and then a thin layer of gold was coated on the samples prior to measurement to improve the conductivity. SEM micrographs were obtained to observe the surface morphologies of different samples.
3. Results and discussion
3.1. Preparation of cellulose esters by MASPS
Cellulose possesses highly-ordered and stable crystalline structure ascribed to strong inter- and intramolecular hydrogen bonds, which make the hydroxyl groups of cellulose difficult to contact and react with chemical reagents, leading to low reaction efficiency in traditional heterogeneous conditions. For the preparation of LCCE in solid phase conditions, esterification reaction hardly takes place without any assisted means because of the poor accessibility of cellulose and the steric hindrance of long chain fatty acids. With combining MA and SPS in the same equipment, the active hydroxyl groups generated from the destruction of strong hydrogen bonds and stable crystalline structure of cellulose induced by intense milling can directly contact and react with esterifying agents. Moreover, the steric effect of long chain fatty acids also can be weakened, attributing to the enhancement of diffusivity and mobility of long chain fatty acids and the improvement of contact between reactants under intense mechanical actions.28 However, the extremely low reactivity of long chain fatty acids toward hydroxyl groups of cellulose also hinders the esterification reaction. With the use of acetic anhydride as co-reactant, long chain fatty acids can be transformed into reactive entities, which quickly react with cellulose. Thus, the reactivity of long chain fatty acids can be effectively improved by the use of co-reactant.
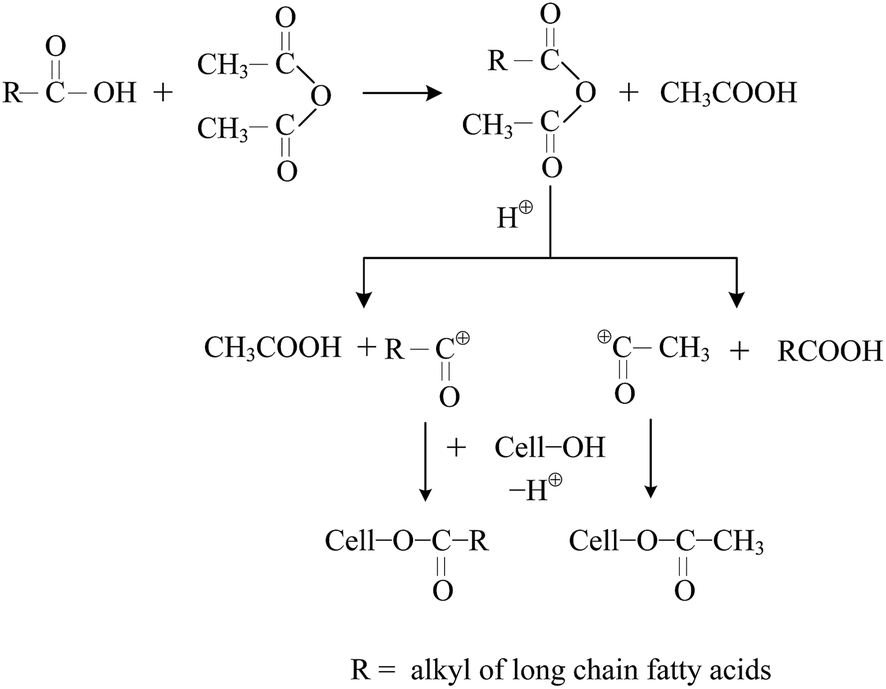
The mechanism of reaction for mixed esterification is illustrated in Fig. 1.20,29 Acetic anhydride and long chain fatty acids react to form mixed acetic-long chain fatty anhydrides. Mixed anhydrides can dissociate in the presence of an acid catalyst to form the corresponding acylium ions, which account for the esterification of cellulose. Two paths are possible. Both the acetylium ions and long chain fatty acylium ions can react with the hydroxyl groups of cellulose. In the previous exploratory experiments, it was found that long chain fatty acids hardly reacted with cellulose without the addition of acetic anhydride, and the DS values of the products were very low and difficult to be determined. This indicates that the mixed esterification technique did not consist of two independent acylation reactions, and acetic anhydride co-reactant influenced the grafting of most fatty chains. On the other hand, Peydecastaing et al. investigated the preparation of cellulose mixed esters by SPS with long chain fatty acids as esterifying agents and acetic anhydride as co-reactant, but the DS values of the products were very low without applying other assisted means, and just about 2% of the hydroxyl groups in cellulose had been esterified.30 Therefore, only the use of MA or co-reactant could not produce the cellulose esters with high DS values by SPS. In this study, co-reactant and MA were simultaneously used as assisted means to enhance the esterification of cellulose with long chain fatty acids by SPS.
 |
| | Fig. 1 Mechanism scheme of the mixed esterification of cellulose with fatty acids and acetic anhydride. | |
After the MASPS of cellulose esters with acetic anhydride as co-reactant, both the acetyl and long chain fatty acyl substituents in esterified cellulose were identified and quantitated by 1H NMR spectroscopy, and the 1H NMR spectra of cellulose acetate–alkanoates with different milling time are presented in Fig. 2 and S1–S14.† The DS values of the grafted fatty acyl chain (DSf) and of the acetyl chain (DSa) were calculated by integrating the characteristic signals of methyl protons of acetyl and fatty acyl chains and cellulose backbone protons, and the results are shown in Table 1. It indicates that the cellulose mixed esters with relatively high values of DSf and total DS were successfully produced. The acetylation reaction was favored when the long chain fatty acids with lower reactivity were used as esterifying agents. It shows that no obvious relationship between the DSa and milling time, which indicates that the acetylation reaction quickly took place when the mixed anhydride or acetic anhydride contacted with cellulose, ascribing to the high reactivity of short chain reagents and the improved contact between cellulose and reagents under intense milling. In contrast, the DSf mainly decreased with the increase of the chain length of fatty acids, and increased as increasing the milling time. The phenomenon that lower DSf was obtained with an increase in carbon atoms of esterifying agents can be explained by two factors: (1) the diffusion of fatty acids into the cellulose is more difficult with an increase in the molecular volume of the acyl groups; (2) when carbon chains are longer, the inductive effect of acyl groups becomes greater, leading to the decrease in the reactivity of fatty acids.4 Fortunately, intense milling could effectively weaken the steric hindrance of long chain fatty acids and enhance the contact between esterifying agents and the hydroxyl groups of cellulose, contributing to the increase in the reactivity of fatty acids. As a result, the DSf values increased with increasing the milling time. Furthermore, it also can be observed that the DSf and DSa of the products gradually decreased when the milling time was unceasingly prolonged. This is because that when the esterification came to a certain extent, the degradation rate of cellulose esters was faster than the esterification rate, leading to the decrease in DS values of the products.31 For the preparation of cellulose esters with high values of DSf and total DS, 120 min can be considered as the optimum milling time.
 |
| | Fig. 2 1H NMR spectrum of cellulose acetate–octanoate (milling time = 30 min) in CDCl3. | |
Table 1 DS values of the cellulose mixed esters calculated from 1H NMR spectra
| Fatty acid |
Milling time (min) |
DSf |
DSa |
DStotal |
| Octanoic acid |
30 |
0.217 |
1.825 |
2.042 |
| 60 |
0.272 |
1.831 |
2.013 |
| 90 |
0.334 |
1.885 |
2.219 |
| 120 |
0.402 |
2.006 |
2.408 |
| 150 |
0.383 |
1.927 |
2.310 |
| Lauric acid |
30 |
0.108 |
1.732 |
1.840 |
| 60 |
0.189 |
1.968 |
2.057 |
| 90 |
0.201 |
2.034 |
2.235 |
| 120 |
0.236 |
2.126 |
2.362 |
| 150 |
0.254 |
1.972 |
2.226 |
| Palmitic acid |
30 |
0.076 |
1.998 |
2.074 |
| 60 |
0.096 |
2.080 |
2.176 |
| 90 |
0.134 |
2.214 |
2.348 |
| 120 |
0.167 |
2.163 |
2.330 |
| 150 |
0.146 |
2.128 |
2.274 |
3.2. Chemical structure by 13C NMR analysis
The chemical structure of the three cellulose mixed esters with the milling time of 120 min was investigated by 13C NMR spectroscopy, and the spectra are presented in Fig. 3 and S15–S16.† As shown in Fig. 3 for the cellulose acetate–octanoate sample, the C-8 carbon atom gives a signal at 31.7 ppm, and peaks at 14.2 and 22.7 ppm correspond to the C-14 and C-15 carbon atoms, respectively. The signals between 20 to 21.5 ppm can be attributed to C-9–13 carbons. Regarding the AGU backbone carbons, the C-6, C-2,3,5, C-4, and C-1 carbons exhibit signals at 62.1, 71.9–72.9, 76.2, and 100.6 ppm, respectively. The most important downfield signals corresponding to the ester carbonyl group (C-7) are shown at 169.4–170.4 ppm, which confirm the acylation of cellulose. In the 13C NMR spectra of cellulose esters, the signals at 170.4, 169.9, and 169.4 ppm correspond to carbonyl carbon at C-6, C-3, and C-2, respectively. Commonly, the distribution of substituents is preferred at C-6, and the order of reactivity for the three hydroxyl groups is C-6–OH ≫ C-3–OH ≈ C-2–OH.32 As calculating from the integration of the corresponding 13C NMR spectra, it was found that the partial DS of C-6:C-3:C-2 in cellulose acetate–octanoate, cellulose acetate–laurate, and cellulose acetate–palmitate were 1:0.91:0.89, 1:0.96:0.91, and 1:0.99:0.96, respectively. These indicate that the distribution of substituents in MASPS produced cellulose esters was more uniform than common esterification methods, and the uniformity increased with the decrease in DSf. During the process of MASPS, intense milling could significantly destroy the hydrogen bonds and thus increase the reactivity of all these three hydroxyl groups in the AGU of cellulose. Furthermore, intense milling could effectively improve the contact between reagents and the lower reactive C-3–OH or C-2–OH, leading to the uniformity of the substituents in cellulose. However, the long chain fatty acylium ions preferred to react with C-6–OH, so the cellulose mixed ester with higher DSf (cellulose acetate–octanoate) had relatively more substituents in C-6.
 |
| | Fig. 3 13C NMR spectrum of cellulose acetate–octanoate (milling time = 120 min) in CDCl3. | |
3.3. Chemical structure by FTIR analysis
The functional groups and chemical structure of untreated cellulose and cellulose esters were examined by FTIR, which is an ideal technique for determining relative efficiency of acylation. The FTIR spectra of MCC before and after esterification by MASPS are presented in Fig. 4. The spectrum of MCC exhibits a strong broad band centered at 3422 cm−1 and a peak at 1636 cm−1, which are attributed to O–H stretching vibration (hydroxyl groups of cellulose) and H–O–H bending of adsorbed water, respectively. The acylation reaction between reagents and the hydroxyl groups of cellulose is clearly indicated by the decreasing intensity of the wide peak at 3422 cm−1, and the greater decrease of this peak implies the more esterification of cellulose. In addition, it can be observed that the peak of O–H stretching vibration shifted to a higher wavenumber after the esterification of cellulose by MASPS, indicating that the hydrogen bond energy of esterified cellulose was higher than that of untreated cellulose. This demonstrates that MA could increase the internal energy of cellulose and decrease the stability of hydrogen bonds.23 The most convincing evidence of successful esterification is the appearance of an absorption band at 1750 cm−1 that corresponds to the stretching vibration of the ester carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and the occurrence of a new peak at 2865 cm−1 in the spectra is attributed to characteristic absorption peak of methylene associated with long chain fatty acyl substituents. Moreover, the bands at 1373, 1236, and 1048 cm−1 assigned to C–H bending, C–O–C ester stretching vibration, and C–O stretching in glycosidic linkages respectively also became stronger after the esterification of cellulose by MASPS,13,33 which were resulted from the intense mechanical actions on cellulose macromolecules and the introduction of acyl groups in cellulose.
O), and the occurrence of a new peak at 2865 cm−1 in the spectra is attributed to characteristic absorption peak of methylene associated with long chain fatty acyl substituents. Moreover, the bands at 1373, 1236, and 1048 cm−1 assigned to C–H bending, C–O–C ester stretching vibration, and C–O stretching in glycosidic linkages respectively also became stronger after the esterification of cellulose by MASPS,13,33 which were resulted from the intense mechanical actions on cellulose macromolecules and the introduction of acyl groups in cellulose.
 |
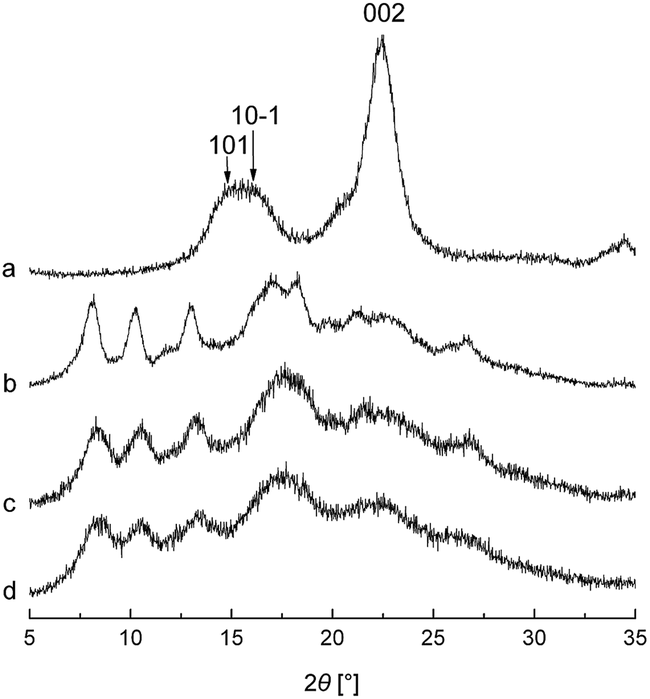
| | Fig. 4 XRD spectra of (a) MCC, (b) cellulose acetate–octanoate (milling time = 120 min), (c) cellulose acetate–laurate (milling time = 120 min), and (d) cellulose acetate–palmitate (milling time = 120 min). | |
3.4. Crystal structure by XRD analysis
XRD measurement is a reliable indicator of the transformation of cellulose structure. Herein it was carried out to prove if MA and chemical modification altered the crystal structure of cellulose. The XRD patterns of MCC and the three synthesized cellulose mixed esters with the milling time of 120 min are shown in Fig. 5. The untreated cellulose sample exhibits three main planes of the crystalline cellulose I structure with the characteristic peaks at around 2θ = 14.8°, 16.2°, and 22.4°, which are assigned to the 101, 10-1, and 002 diffraction planes, respectively.23 After chemical modification by MASPS, the three main planes almost disappeared in the XRD patterns of the three cellulose mixed esters, and some new peaks appeared at around 7°–19° and 26.7°. During the process of MASPS, MA remarkably induced the decrease in crystalline phase, the variation of crystallite size and the distortion of stable crystalline structure of cellulose linked with ball impact and collision, and the crystal structure of cellulose was nearly completely destroyed after the milling time of 120 min. When subjected to intense milling, the destruction of stable hydrogen bonds and crystal structure of cellulose could result in the generation of active and free hydroxy groups, which help to improve the reactivity of cellulose and increase the DS of cellulose esters.31 Even though intense milling reduced the crystallinity of cellulose for the esterification of cellulose by MASPS, the grafting of long chain fatty acyls on cellulose could make the crystallization of these long side chains. So, the new peaks appearing in the XRD patterns of esterified cellulose could ascribe to the formation of new ordered structures associated with the crystallization of long aliphatic side chains. Evidently, the XRD pattern of cellulose acetate–octanoate with higher DSf value shows greater diffraction intensity in the new peaks.4
 |
| | Fig. 5 FTIR spectra of (a) MCC, (b) cellulose acetate–octanoate (milling time = 120 min), (c) cellulose acetate–laurate (milling time = 120 min), and (d) cellulose acetate–palmitate (milling time = 120 min). | |
3.5. Morphology observation by SEM analysis
The surface morphologies of different samples can be clearly observed from SEM micrographs, which are illustrated in Fig. 6. The untreated MCC consists of short cellulose bundles with smooth surfaces, which can be seen in Fig. 6a. After chemical modification, the large particles with rough surface were obtained for all the three cellulose mixed esters with the milling time of 120 min (Fig. 6b–d). In the process of MASPS, the cellulose bundles were remarkably damaged and fractured, contributing to the generation of small irregular particles with rough and fresh surfaces. Due to the introduction of acyl substituents in cellulose and the physical forces of ball milling, these small particles easily agglomerated to form large particles. The destruction of fibrous structure and increase of specific surface area could provide a facile access for esterifying agents to contact with the hydroxy groups of cellulose, which increased the chemical reactivity of cellulose and thus enhanced its esterification reaction. Furthermore, the corrosion occurred as penetration or rupture from the granules surface to core during MASPS, indicating that the internal crystalline region of cellulose was also damaged by ball milling to form an amorphous region, and the esterification reaction took place not only in the surface amorphous region but also in the internal damaged region.34 This is because the destruction of compact surface coatings and crystal structure of cellulose induced by intense milling built new paths for esterifying agents to enter into the interior of cellulose, which led to the direct synthesis of cellulose esters under solid phase conditions. It also can be observed that the adhesion and agglomeration of the esterified cellulose particles are more obvious with the longer chain fatty acyl substituents. This may due to that cellulose macromolecules with longer alkyl side chains could easily interact with each other and thus led to the adhesion and agglomeration of the cellulose particles.
 |
| | Fig. 6 SEM micrographs of (a) MCC, (b) cellulose acetate–octanoate (milling time = 120 min), (c) cellulose acetate–laurate (milling time = 120 min), and (d) cellulose acetate–palmitate (milling time = 120 min). | |
4. Conclusion
A novel technology was developed for the synthesis of cellulose esters with long chain substituents. It was performed in a stirring ball milling under solid phase condition with acetic anhydride as co-reactant, and octanoic acid, lauric acid, and palmitic acid as long chain esterifying agents. 1H and 13C NMR spectra confirmed that long chain fatty acyl groups were successfully grafted on cellulose by MASPS, ascribing to the improved reactivity of long chain fatty acids induced by the use of acetic anhydride as co-reactant and the combination of MA and esterification in the same equipment. In addition, acetyl groups were quickly grafted on cellulose, resulting from the high reactivity of acetylium ion and the improved contact between cellulose and reagents under intense milling. Due to the difficult diffusion of fatty acid into cellulose and the inductive effect of acyl groups, the reactivity of fatty acids and the DSf values decreased with the increase in their chain length. The distribution of substituents in MASPS produced cellulose esters was more uniform than common esterification methods, resulting from the increase in the reactivity of all these three hydroxyl groups in the AGU of cellulose induced by MA. In addition, the changes in chemical structure, crystal structure, and surface morphology of the cellulose before and after esterification by MASPS were demonstrated by FTIR, XRD, and SEM measurements, respectively. Further investigations for the specific properties and applications of these MASPS produced cellulose mixed esters will be carried out in our next works. Hopefully, this green, simple, and efficient method for the production of cellulose esters with long chain substituents has significantly potential application for industrial production.
Acknowledgements
This research was supported by National Natural Science Foundation of China (no. 51163002), Guangxi Natural Science Foundation of China (no. 2013GXNSFDA019004 and no. 2013GXNSFBA019028), Guangxi Distinguished Experts Special Foundation of China, and the Scientific Research Foundation of Guangxi University, China (Grant no. XTZ140787).
References
- D. Klemm, B. Heublein, H. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- B. R. Caes, M. J. Palte and R. T. Raines, Chem. Sci., 2013, 4, 196–199 RSC.
- B. Medronho, A. Romano, M. G. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581–587 CrossRef CAS.
- U. Ratanakamnuan, D. Atong and D. Aht-Ong, Carbohydr. Polym., 2012, 87, 84–94 CrossRef CAS PubMed.
- S. R. Labafzadeh, J. S. Kavakka, K. Vyavaharkar, K. Sievänen and I. Kilpeläinen, RSC Adv., 2014, 4, 22434–22441 RSC.
- T. Ema, T. Komiyama, S. Sunami and T. Sakai, RSC Adv., 2014, 4, 2523–2525 RSC.
- S. C. Fox, B. Li, D. Xu and K. J. Edgar, Biomacromolecules, 2011, 12, 1956–1972 CrossRef CAS PubMed.
- A. Chandran, S. Kuriakose and T. Mathew, Polym. Adv. Technol., 2013, 24, 525–531 CrossRef CAS.
- K. J. Edgar, C. M. Buchanan, J. S. Debenham, P. A. Rundquist, B. D. Seiler, M. C. Shelton and D. Tindall, Prog. Polym. Sci., 2001, 26, 1605–1688 CrossRef CAS.
- L. Crépy, L. Chaveriat, J. Banoub, P. Martin and N. Joly, ChemSusChem, 2009, 2, 165–170 CrossRef PubMed.
- L. Crépy, V. Miri, N. Joly, P. Martin and J. Lefebvre, Carbohydr. Polym., 2011, 83, 1812–1820 CrossRef PubMed.
- Y. Xu, C. Wang, N. M. Stark, Z. Cai and F. Chu, Carbohydr. Polym., 2012, 88, 422–427 CrossRef CAS PubMed.
- F. Xu, J. Jiang, R. Sun, D. She, B. Peng, J. Sun and J. F. Kennedy, Carbohydr. Polym., 2008, 73, 612–620 CrossRef CAS PubMed.
- S. Boufi and M. N. Belgacem, Cellulose, 2006, 13, 81–94 CrossRef CAS.
- M. A. Hussain, T. Liebert and T. Heinze, Macromol. Rapid Commun., 2004, 25, 916–920 CrossRef CAS.
- P. Blaney, R. Grigg and V. Sridharan, Chem. Rev., 2002, 102, 2607–2624 CrossRef CAS PubMed.
- A. Loupy, C. R. Chim., 2004, 7, 103–112 CrossRef CAS PubMed.
- Y. Chang, C. Liu, C. Guo, Y. Wang, J. Fang and W. Cheng, J. Org. Chem., 2008, 73, 7197–7203 CrossRef CAS PubMed.
- A. T. W. M. Hendriks and G. Zeeman, Bioresour. Technol., 2009, 100, 10–18 CrossRef CAS PubMed.
- C. Vaca-Garcia, S. Thiebaud, M. E. Borredon and G. Gozzelino, J. Am. Oil Chem. Soc., 1998, 75, 315–319 CrossRef CAS PubMed.
- C. Sasikumar, D. S. Rao, S. Srikanth, N. K. Mukhopadhyay and S. P. Mehrotra, Hydrometallurgy, 2007, 88, 154–169 CrossRef CAS PubMed.
- C. Chen, Y. Shen and A. Yeh, J. Agric. Food Chem., 2010, 58, 9083–9091 CrossRef CAS PubMed.
- Z. Liao, Z. Huang, H. Hu, Y. Zhang and Y. Tan, Bioresour. Technol., 2011, 102, 7953–7958 CrossRef CAS PubMed.
- Z. Huang, X. Liang, H. Hu, L. Gao, Y. Chen and Z. Tong, Polym. Degrad. Stab., 2009, 94, 1737–1745 CrossRef CAS PubMed.
- P. Baláž and M. Achimovičová, Hydrometallurgy, 2006, 84, 60–68 CrossRef PubMed.
- Z. Huang, X. Xie, Y. Chen, J. Lu and Z. Tong, C. R. Chim., 2008, 11, 73–79 CrossRef CAS PubMed.
- C. Satgé, R. Granet, B. Verneuil, P. Branland and P. Krausz, C. R. Chim., 2004, 7, 135–142 CrossRef PubMed.
- Y. Zhang, T. Gan, H. Hu, Z. Huang, A. Huang, Y. Zhu, Z. Feng and M. Yang, Ind. Eng. Chem. Res., 2014, 53, 2114–2120 CrossRef.
- C. Vaca-Garcia and M. E. Borredon, Bioresour. Technol., 1999, 70, 135–142 CrossRef CAS.
- J. Peydecastaing, C. Vaca-Garcia and E. Borredon, Cellulose, 2011, 18, 1015–1021 CrossRef CAS PubMed.
- Y. Zhang, T. Gan, Y. Luo, X. Zhao, H. Hu, Z. Huang, A. Huang and X. Qin, Compos. Sci. Technol., 2014, 102, 139–144 CrossRef CAS PubMed.
- J. Chen, J. Zhang, Y. Feng, J. Wu, J. He and J. Zhang, J. Membr. Sci., 2014, 469, 507–514 CrossRef CAS PubMed.
- J. M. Fang, R. Sun, P. Fowler, J. Tomkinson and C. A. S. Hill, J. Appl. Polym. Sci., 1999, 74, 2301–2311 CrossRef CAS.
- Z. Huang, Y. Tan, Y. Zhang, X. Liu, H. Hu, Y. Qin and H. Huang, Bioresour. Technol., 2012, 118, 624–627 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra02393a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.