
Magnetic solid phase extraction of lead(II) and cadmium(II) on a magnetic phosphorus-containing polymer (M-PhCP) for their microsampling flame atomic absorption spectrometric determinations
E. Yilmaza,
R. M. Alosmanovb and
M. Soylak*a
aErciyes University, Fen Faculty, Department of Chemistry, 38039 Kayseri, Turkey. E-mail: soylak@erciyes.edu.tr; Fax: +90 352 4374933; Tel: +90 352 4374933
bBaku State University, Chemistry Department, Z. Khalilov str., 23, Baku, AZ1148, Azerbaijan
First published on 9th March 2015
Abstract
A magnetic solid-phase extraction (M-SPE) procedure on a magnetic phosphorus-containing polymer (M-PhCP) was established for the separation/preconcentration of trace amounts of lead and cadmium in water samples prior to their microsampling flame atomic absorption spectrometric determination. The separation of lead(II) and cadmium(II) adsorbed as 2-(5-bromo-2-pyridylazo)-5-diethylamino-phenol (5-Br-PADAP) chelates on M-PhCP from aqueous solution was simply achieved by applying an external magnetic field via a permanent magnet. The important analytical factors governing the extraction efficiency such as pH, amount of adsorbent, eluent concentration and volume, vortex time, ligand volume, and sample volume were investigated and optimized. The influences of matrix components were also investigated. The limit of detection (LOD) of Pb and Cd was 2.7 and 1.1 μg L−1, respectively. The accuracy of the proposed method was proved by analysis of the TMDA-64.2 Lake Ontario water and SPS-WW2 waste water certified reference materials and addition–recovery tests. The method was successfully applied for the determination of lead and cadmium in water samples.
1. Introduction
Environmental exposure to heavy metals is a well known risk factor for human health and is also harmful to plants and animals.1–5 Heavy metals like lead and cadmium are extremely toxic even at very low levels to humans and animals, while metals such as copper, nickel and zinc play important roles in several biochemical processes and their compounds have bactericidal activity.6–9 However, if they are in excess, these metals can cause various chronic disorders in human beings.10 Environmental samples are considered as a main source of human exposure to lead and cadmium. Therefore, the monitoring of lead and cadmium in environmental samples is very important to ensure the quality of human health.11–13Due to the very low concentrations of heavy metals in real samples, their determinations demand very sensitive analytical techniques including inductively coupled plasma mass spectrometry (ICP-MS), electrothermal atomic absorption spectrometry (ET-AAS), inductively coupled plasma atomic emission spectrometry (ICP-AES) and X-ray fluorescence (XRF).14–16 However, when compared to flame atomic absorption spectrometry (FAAS), the available analytical techniques have some disadvantages such as high cost, slowness and greater proneness to matrix interferences, as well as their high sensitivity advantages.16,17 FAAS is, in principle, a suitable technique due to the low cost and easy operation. However, FAAS does not have sufficient sensitivity and selectivity for the determination of heavy metals in real samples because of their low concentration, which may be lower than the detection limit of FAAS, and matrix effects.18,19
A separation/preconcentration procedure is generally required to solve sensitivity and selectivity problems. Several techniques have been developed for the separation and preconcentration of trace metals including: cloud point extraction,20 liquid–liquid extraction,21 precipitation/co-precipitation,13 and solid-phase extraction (SPE).17 SPE is considered superior to other techniques due to its simplicity, the consumption of small volumes of organic solvent, and the ability to obtain a higher preconcentration factor and greater speed.17,22,23
Magnetic adsorbents have attracted much attention because of their unique magnetic properties for SPE procedures.24–27 In a magnetic SPE procedure, a magnetic sorbent is added to the sample solution containing the target analyte. The analytes are adsorbed onto the magnetic adsorbent whether under mixing or standing conditions. The adsorbent with the captured analytes is then isolated from the sample solution by using an appropriate magnet. Finally, the target analytes adsorbed on the adsorbents are desorbed with suitable elution and analyzed using an appropriate method.28,29 The application of magnetic SPE simplifies the preconcentration/separation steps for analytes.28,29 The magnetic adsorbents do not need to be packed into a cartridge or column, as in traditional SPE procedures, and the phase separation can be realized easily by applying an external magnetic field.
In the present paper, a magnetic phosphorus-containing polymeric sorbent (M-PhCP) was used for the magnetic solid phase extraction (M-SPE) of lead(II) and cadmium(II) as 2-(5-bromo-2-pyridylazo)-5-diethylamino-phenol (5-Br-PADAP) chelates from natural water samples prior to their microsampling flame atomic absorption spectrometric determination.
2. Experimental
2.1. Chemicals and solutions
The water used in the experiments was ultrapure water which was prepared using a milli-Q system (18.2 MΩ cm) from Millipore (Bedford, MA, USA). All chemicals used were analytical reagent grade and were used without further purification. The analytical-grade acids and other chemicals used in this study were purchased from Merck, Darmstadt, Germany. The chelating agent, 2-(5-bromo-2-pyridylazo)-5-diethylamino-phenol (5-Br-PADAP) from Sigma-Aldrich (St. Louis, MO, USA), was prepared daily in ethanol as a 0.1% (m/v) solution. The working metal ion solutions used for the experiments were prepared by the dilution of 1000 mg L−1 of stock solutions.Buffer solutions were prepared by using a combination of salts and solutions as follows: phosphate buffer solution (pH 2.0–4.0, sodium dihydrogen phosphate/phosphoric acid) and phosphate buffer solution (pH 5.0–7.0 sodium dihydrogen phosphate/disodium hydrogen phosphate). TMDA-64.2 Lake Ontario water (National Water Research Institute, Ontario, Canada) and SPS-WW2 waste water (Spectrapure Standards AS, Oslo, Norway) certified reference materials were used.
2.2. Apparatus
A Perkin-Elmer Model 3110 (Norwalk, CT, USA) flame atomic absorption spectrometer equipped with a hollow cathode lamp and an air/acetylene flame as an atomizer was used for the measurements. The continuous aspiration mode was used to measure the metal ion concentrations in the eluate. A hundred μL of the eluate phase was injected into the FAAS nebulizer using a home-made micro-sample introduction system consisting of a Teflon funnel and an Eppendorf pipette and the peak height was measured.11,30The FT-IR spectra of the phosphorus-containing polymeric sorbent (PhCPS) and magnetic phosphorus-containing polymeric sorbent (M-PhCP) were recorded on a Perkin-Elmer Spectrum 400 ATR-FT-IR spectrometer (Waltham, MA, USA). Scanning electron microscopy (SEM) images were obtained on a LEO 440 scanning electron microscope with an accelerating voltage of 20 kV. For the SEM measurements, the samples were covered with Au/Pd. X-ray diffractions (XRD) pattern were taken with Bruker AXS D8 advanced diffractometer. The surface area, pore volume and pore width of the magnetic phosphorus-containing polymeric sorbent (M-PhCP) were determined by the BET-N2 method using a Micromeritics Gemini VII analyzer. A pH meter, Sartorious PT-10 (Germany) was used for measuring the pH values. A VWR international model (Germany) vortex mixer was used during the experimental period.
2.3. Synthesis of the magnetic adsorbent
A phosphorus-containing polymeric sorbent was used as a matrix for the magnetic adsorbent (PhCPS). PhCPS was obtained as described in ref. 31. A 5% solution of polybutadiene in CCl4 was subjected to oxidative chlorophosphorylation by PCl3 under the action of oxygen. This resulted in the formation of a modified substance with a three-dimensional structure containing functional groups with P–Cl bonds, which were further processed with water for hydrolysis. The adsorbent was purified and dried at 50 °C in a vacuum cabinet.31 Its structure was studied by NMR in the solid phase.32 It was established that the PhCPS has a three-dimensional structure (crosslinked polymers) and contains phosphonate and phosphate groups. The magnetic adsorbent was prepared as follows: at first, ferric and ferrous chlorides (molar ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were dissolved in water at a concentration of 0.3 M iron ions. Then, this mixture was added to the PhCPS. The volume of the solution was 100 mL and the amount of PhCPS was 1.0 g. The suspension was stirred for two hours. Then, the PhPCS was separated from the aqueous solution and dried in air. Then, 50 mL of sodium hydroxide of a concentration of 4 M was added to the air-dried adsorbents. Argon gas was passed into the mixture and the reaction was allowed to proceed for 1 hour at 80 °C under constant and vigorous stirring in reflux equipment. The obtained pure black magnetic phosphorus-containing polymeric sorbent (M-PhCP) was separated using a magnet, washed several times with water and ethanol, and then finally dried in a vacuum oven at 50 °C.
:1) were dissolved in water at a concentration of 0.3 M iron ions. Then, this mixture was added to the PhCPS. The volume of the solution was 100 mL and the amount of PhCPS was 1.0 g. The suspension was stirred for two hours. Then, the PhPCS was separated from the aqueous solution and dried in air. Then, 50 mL of sodium hydroxide of a concentration of 4 M was added to the air-dried adsorbents. Argon gas was passed into the mixture and the reaction was allowed to proceed for 1 hour at 80 °C under constant and vigorous stirring in reflux equipment. The obtained pure black magnetic phosphorus-containing polymeric sorbent (M-PhCP) was separated using a magnet, washed several times with water and ethanol, and then finally dried in a vacuum oven at 50 °C.
2.4. General procedure
25 mL of model solutions containing 20 μg lead(II) and 10 μg cadmium(II) ions, 100 μL of 0.1% 5-Br-PADAP and 5 mL phosphate buffer were transferred to 50 mL conical-bottom glass centrifuge tubes and the pH of the solutions was adjusted to pH 6.0. Then, 100 mg of the magnetic adsorbent was added, and the solution was mixed using a vortex agitator for 3 min to facilitate the adsorption of the metal ions onto the adsorbent. A neodymium magnet was subsequently held around the test tube for the easy and quick collection of the magnetic adsorbent at the bottom of the test tube. Then, the supernatants were decanted directly. 5.0 mL of a 0.5 mol L−1 HNO3 in acetone solution was added as the eluate and it was mixed again for 1 min. The magnetic adsorbent was collected from the eluate with the magnet. Then, the eluate was transferred into another tube using a pipette. The acetone in the eluate was evaporated till approximately 500 μL remained. The final volume was completed to 500 μL with acetone in a HPLC vial of 500 μL volume. Finally, 100 μL of the eluate was measured using a micropipette and injected into the microinjection system coupled with a FAAS nebulizer. The peak height in continuous aspiration mode was measured.2.5. Analysis of real samples
River water and dam water from Kayseri, waste water samples from plants located in the Kayseri Organized Industrial Area and well water from Ankara, Turkey were collected. The samples used were collected in pre-washed polyethylene containers and were filtered through a Millipore cellulose membrane filter (0.45 mm of pore size). Then, the developed magnetic SPE method was applied to the water samples.The developed magnetic SPE method was also applied to TMDA 64.2 Lake Ontario water and SPS-WW2 waste water certified reference materials. The analytes in the eluate were determined using flame AAS and a microsampling system.
3. Results and discussion
3.1. Characterization of magnetic adsorbent
The FT-IR spectra of the phosphorus-containing polymeric sorbent (PhCPS) and magnetic phosphorus-containing polymeric sorbent (M-PhCP) (A and B) are shown in Fig. 1. In the IR spectrum of PhCPS (A), the typical bands can be assigned as follows: O–H: 3343 cm−1, C–H: 2978 cm−1, 2935 cm−1, O–H (P–O–H): 2862 cm−1, 2494 cm−1 P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (resonance state): 1622 cm−1, PO: 1183 cm−1, C–O (P–C–O): 1050 cm−1 and C–Cl: 495 cm−1, respectively. In the IR spectrum of M-PhCP (B), the sorption peak of OH for PhCPS had been remarkably weakened after the magnetizing process because of the change of hydrogen to iron groups. Fe–O has a characteristic band at 546 cm−1 (ref. 33) and this also indicates that Fe3O4 molecules were successfully formed within the structure of the phosphorus-containing polymeric sorbent.
O (resonance state): 1622 cm−1, PO: 1183 cm−1, C–O (P–C–O): 1050 cm−1 and C–Cl: 495 cm−1, respectively. In the IR spectrum of M-PhCP (B), the sorption peak of OH for PhCPS had been remarkably weakened after the magnetizing process because of the change of hydrogen to iron groups. Fe–O has a characteristic band at 546 cm−1 (ref. 33) and this also indicates that Fe3O4 molecules were successfully formed within the structure of the phosphorus-containing polymeric sorbent.
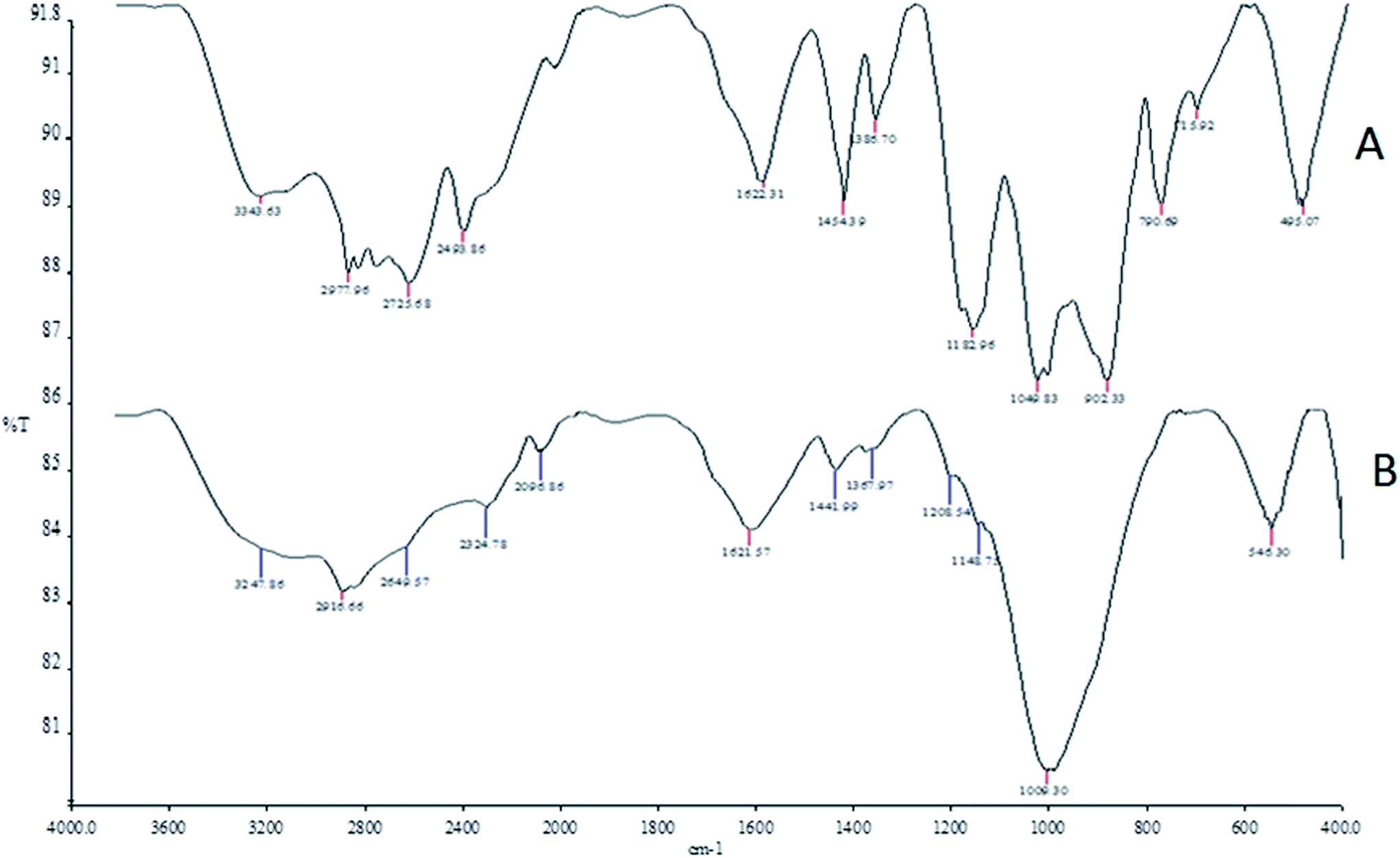 | ||
| Fig. 1 The FT-IR spectra of the phosphorus-containing polymeric sorbent (PhCPS) and magnetic phosphorus-containing polymeric sorbent (M-PhCP). | ||
Scanning electron microscopy (SEM) micrographs presented in Fig. 2 show the porous surface structures of the PhCPS (A) and M-PhCP (B). There are some differences between the morphology of the PhCPS and the M-PhCP. The PhCPS surface is quite rough, providing a large exposed surface area for the adsorption of metal–ligand complexes. To investigate, the presence and distribution of magnetic particles in the magnetic phosphorus-containing polymer, the elemental mapping image (EMI) was carried out using SEM-EDX analysis. For this purpose, the elemental mapping image of the iron element was performed (Fig. 3). The obtained elemental mapping image indicates that the magnetic iron particles are homogeneously dispersed on the phosphorus-containing polymer. The elemental composition of the magnetic phosphorus-containing polymer was also analyzed using SEM-EDX. The sample was covered with Au. The obtained spectrum is given in Fig. 4. The amount of iron found was 6.8 wt%.
 | ||
| Fig. 2 SEM images of the phosphorus-containing polymeric sorbent (PhCPS) (A) and magnetic phosphorus-containing polymeric sorbent (M-PhCP) (B). | ||
 | ||
| Fig. 3 Elemental mapping image (EMI) of the magnetic phosphorus-containing polymer. | ||
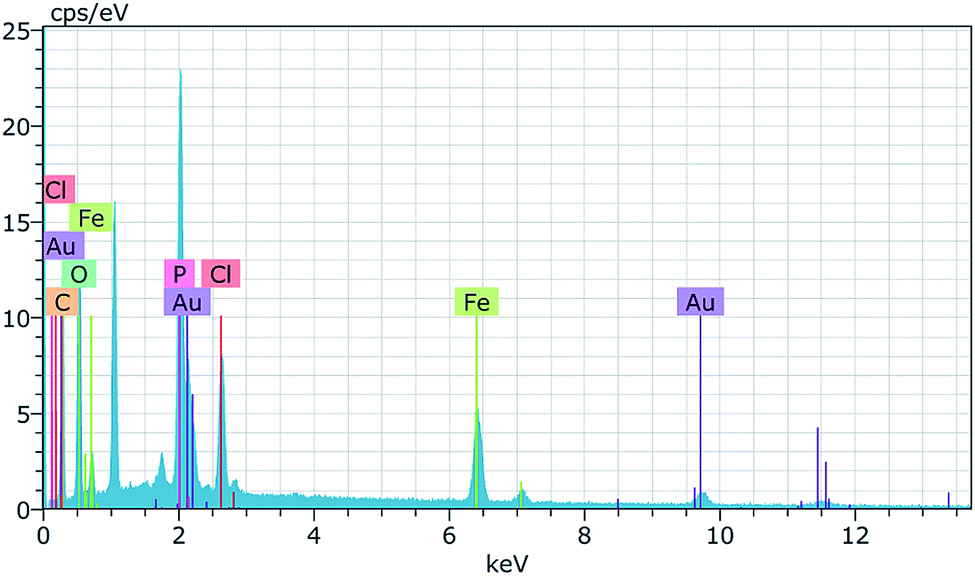 | ||
| Fig. 4 EDX spectrum of the magnetic phosphorus-containing polymer. | ||
The structures of the phosphorus-containing polymer (A) and the magnetic phosphorus-containing polymer (B) were characterized by XRD and the diffractograms are given in Fig. 5. There are four types of iron oxides commonly formed including magnetite (Fe3O4), maghemite (γ-Fe2O3), hematite (α-Fe2O3) and goethite (FeO(OH)). Among them, only magnetite and maghemite are magnetic. The XRD pattern of the magnetic phosphorus-containing polymer displays five main diffraction peaks at 2θ = 31.8°, 34.3°, 45.5°, 55.3° and 62.4° that can be assigned to maghemite or magnetite. Other peaks are also observed at 2θ = 19.1°, 33.3°, 42.2° and 59.5° for the magnetic phosphorus-containing polymer, which may be related to the presence of goethite. The diffraction pattern of the magnetic phosphorus-containing polymer is close to the standard pattern for JCPD. It is clearly seen that the particles do not show sharp diffraction peaks. Instead, a broad band appears. This is typical for amorphous materials and also for ultrafine crystalline materials where diffraction peaks cannot be well resolved.
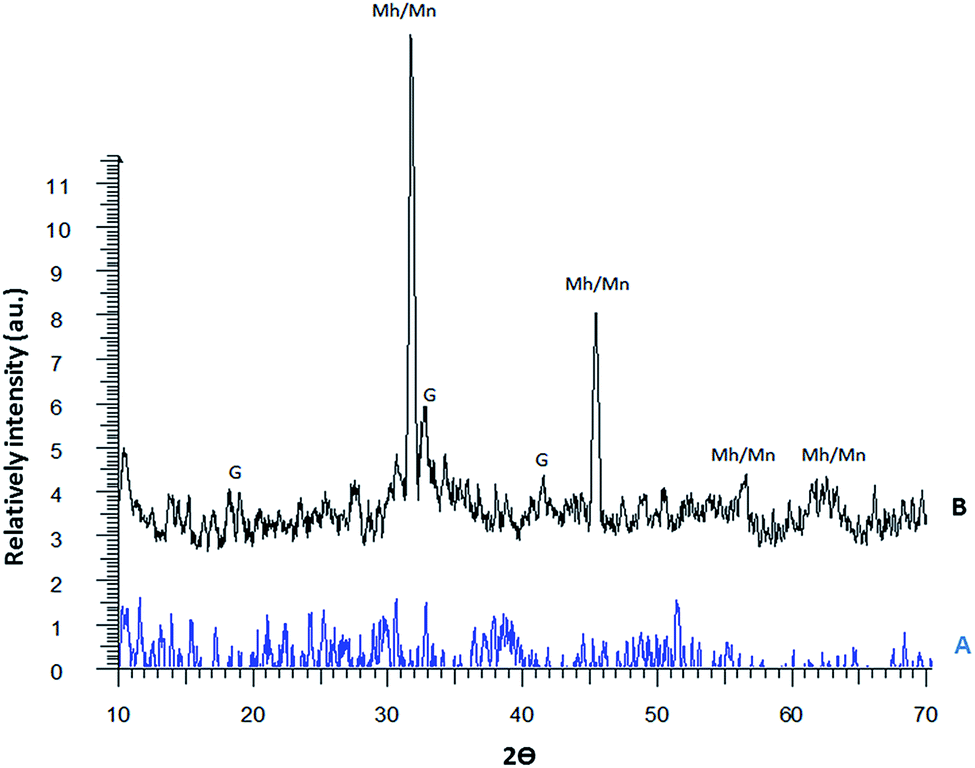 | ||
| Fig. 5 XRD patterns of the phosphorus-containing polymer (A) and the magnetic phosphorus-containing polymer (B). | ||
From the IR spectra it is clear that the typical bands for the phosphorus-containing polymer (O–H, C–H, O–H (P–O–H): PO (resonance state) PO: 1183 cm−1, C–O (P–C–O) and C–Cl: 495 cm−1) also appeared for the magnetic phosphorus-containing polymer. The IR spectrum for the magnetic phosphorus-containing polymer shows that the polymeric parts of the magnetic phosphorus-containing polymer are effective for the adsorption of analytes as their 5-Br-PADAP chelates. Van der Waals interactions are important for adsorption studies. The functional groups of the phosphorus-containing polymer provide effective van der Waals interactions between analyte–5-Br-PADAP chelates and the adsorbent.
The adsorption efficiency is related to the internal surface area and pore volume. The BET surface area, pore volume and pore width of the M-PhCP were found to be 7.53 m2 g−1, 0.00535 cm3 g−1 and 2.84 nm, respectively. The BET isotherm of the M-PhCP sample in Fig. 6 shows that the contribution of mesopores to the total surface area and pore volume is significantly higher than that of macropores.34
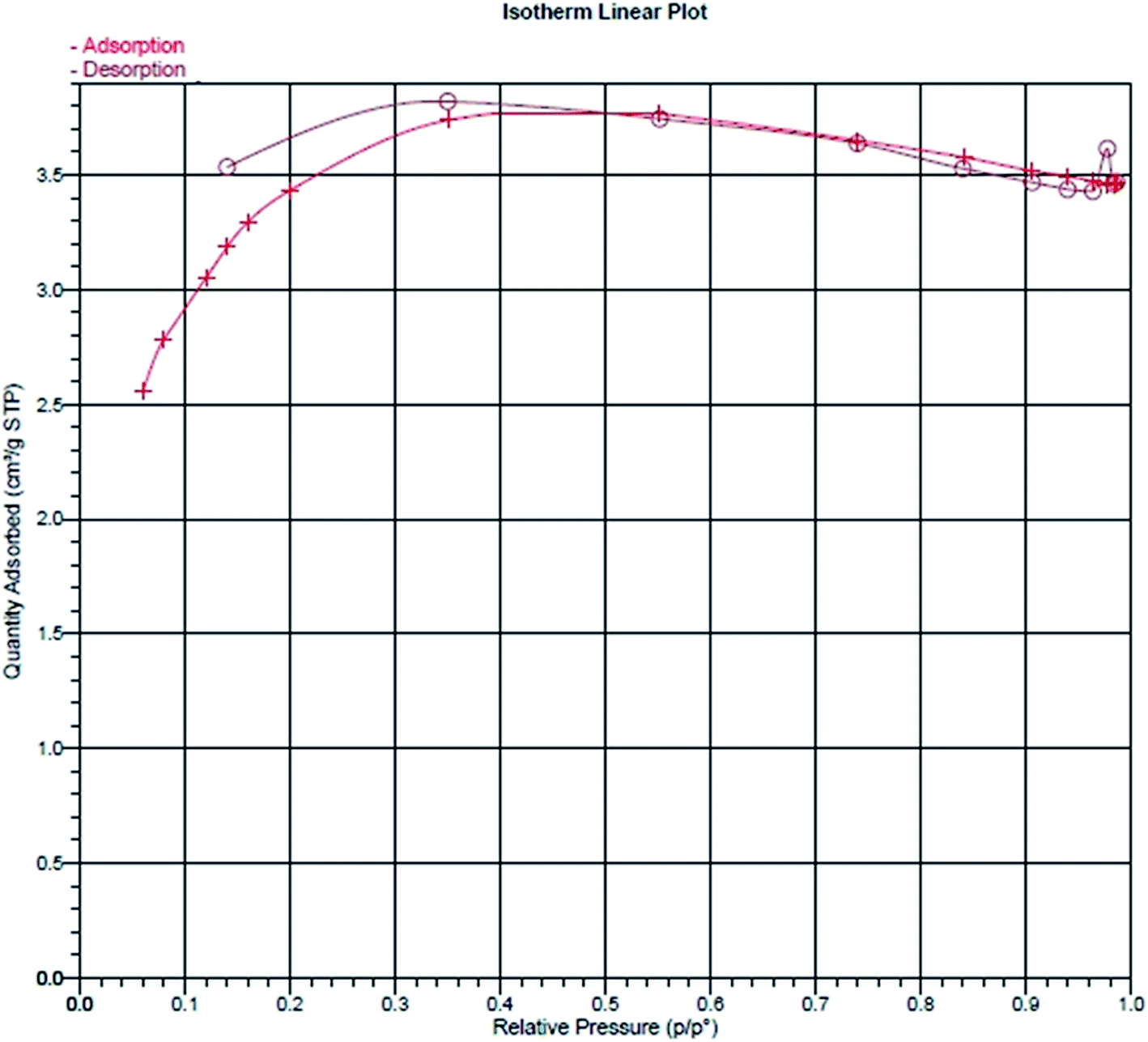 | ||
| Fig. 6 Nitrogen adsorption/desorption isotherm of the magnetic phosphorus-containing polymeric sorbent (M-PhCP). | ||
3.2. Optimization of the extraction parameters
The pH of the sample solution has an important role in extraction studies of metal ions at trace levels.34–39 The pH of model solutions containing 20 μg lead(II) and 10 μg cadmium(II) was studied within the range of 2.0–8.0 using buffer solutions and then the separation/preconcentration procedure described was applied. As can be seen in Fig. 7, quantitative recoveries for analyte ions were obtained at pH 6.0. Hence, pH 6.0 was used in all experiments as the optimum value.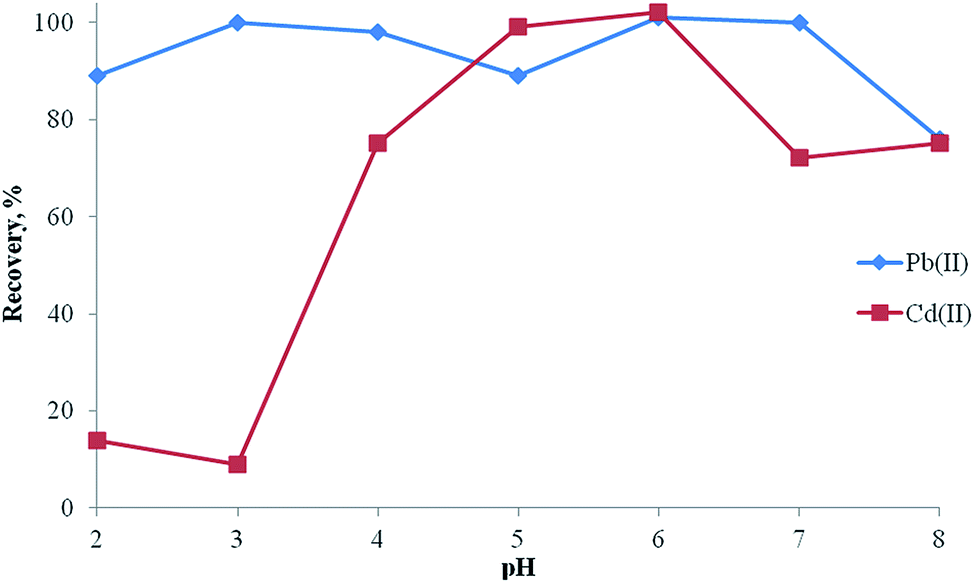 | ||
| Fig. 7 Effect of pH on the recoveries of Pb(II) and Cd(II) (N (number of samples analyzed) = 3, amount of analytes: 20 μg lead(II) and 10 μg cadmium(II), amount of adsorbent: 100 mg, amount of ligand: 0.1 mg, eluent: 5 mL of 0.5 mol L−1 HNO3 in acetone). | ||
Compared to conventional adsorbents, magnetic adsorbents have some advantages including high extraction capacity, rapid extraction dynamics and high extraction efficiency.29,40–44 The amount of the magnetic adsorbent was studied in the range of 25–100 mg of magnetic adsorbent. The recoveries for Pb(II) and Cd(II) were increased and reached quantitative values at 100 mg of magnetic adsorbent. Therefore, 100 mg of adsorbent was selected for further experiments.
The recoveries of the analytes by the iron species only were 75% and the recoveries of the analytes on the phosphorus-containing polymer with a ligand without magnetic properties was lower than 50%. The recoveries of the analytes on the magnetic phosphorus-containing polymer with ligand (presented system) were quantitative (>95%).
The influences of various eluents on the recoveries of Pb(II) and Cd(II) ions from the magnetic phosphorus-containing polymeric sorbent were also examined. The results are summarized in Table 1. It should be pointed out that the regeneration of the magnetic property of the adsorbent is possible due to the dissolution of Fe3O4 in a high acid concentration. Hence, low concentrations of HNO3 were examined in this study. The recoveries of the analyte ions were quantitative with 0.5 M HNO3 in acetone. The recoveries were not quantitative for the other eluents. 0.5 M HNO3 in acetone was used as the eluent in all further works for the quantitative recovery of metal ions from M-PhCP. At the same time, the effect of the eluent volume on the recoveries of Cd(II) and Pb(II) was studied. It was found that quantitative recoveries could be obtained with 5.0 mL of 0.5 M HNO3 in acetone solution and more than 5.0 mL. Therefore, 5.0 mL of eluent was used in the following experiments.
| Eluent type | Recovery, % | |
|---|---|---|
| Pb(II) | Cd(II) | |
| a N: number of samples analyzed. | ||
| 7 mL 0.05 mol L−1 HNO3 in acetone | 77 ± 1 | 69 ± 4 |
| 7 mL 0.1 mol L−1 HNO3 in acetone | 73 ± 6 | 69 ± 3 |
| 7 mL 0.25 mol L−1 HNO3 in acetone | 70 ± 10 | 46 ± 2 |
| 7 mL 0.5 mol L−1 HNO3 in acetone | 95 ± 2 | 99 ± 2 |
| 1 mL 0.5 mol L−1 HNO3 in acetone | 44 ± 0 | 50 ± 0 |
| 2 mL 0.5 mol L−1 HNO3 in acetone | 67 ± 0 | 45 ± 1 |
| 3 mL 0.5 mol L−1 HNO3 in acetone | 104 ± 6 | 45 ± 0 |
| 4 mL 0.5 mol L−1 HNO3 in acetone | 89 ± 2 | 45 ± 0 |
| 5 mL 0.5 mol L−1 HNO3 in acetone | 95 ± 2 | 101 ± 3 |
In order to obtain a hydrophobic metal complex, 5-Br-PADAP was used as the complexing agent. The effects of the amounts of 5-Br-PADAP on the recoveries of the analytes were also examined with model solutions containing 20 μg lead(II) and 10 μg cadmium(II) in the range of 0.0–0.5 mg of 5-Br-PADAP. The results are shown in Fig. 8. The results are not quantitative without the ligand. Quantitative recoveries were obtained using 0.1 mg of 5-Br-PADAP. Hence, 0.1 mg of 5-Br-PADAP was used for further study.
 | ||
| Fig. 8 Effect of the amount of ligand on the recoveries of Pb(II) and Cd(II) (N = 3, amount of analytes: 20 μg lead(II) and 10 μg cadmium(II), pH: 6.0, amount of adsorbent: 100 mg, eluent: 5 mL, 0.5 mol L−1 HNO3 in acetone). | ||
In order to obtain a high preconcentration factor,45–52 the magnetic SPE method was applied to 10, 15, 20, 30 and 40 mL of sample solution. Quantitative recoveries of the analytes could be obtained using 100 mg of the M-PhCP when the sample volume was 40 mL. The preconcentration factor was 80. Therefore, the developed method is very suitable for the preconcentration of trace lead and cadmium from large volumes of sample solution.
The effects of common interfering ions on the recoveries of the Pb(II) and Cd(II) ions were also examined. The obtained results are given in Table 2. The recoveries of all analyte ions were quantitative for the given concentrations of interfering ions. The results demonstrate that thousand fold excesses of the analyte ions have no effect on the preconcentration and determination of the Pb(II) and Cd(II).
| Ions | Concentration, μg mL−1 | Added as | Recovery, % | |
|---|---|---|---|---|
| Pb(II) | Cd(II) | |||
| Na+ | 10000 |
NaNO3 | 100 ± 0 | 99 ± 2 |
| K+ | 10000 |
KCl | 95 ± 4 | 97 ± 1 |
| Co2+ | 25 | Co(NO3)2·6H2O | 98 ± 5 | 101 ± 1 |
| Cu2+ | 25 | Cu(NO3)2·3H2O | 95 ± 2 | 99 ± 2 |
| Zn2+ | 25 | Zn(NO3)2·6H2O | 97 ± 2 | 101 ± 3 |
| Fe3+ | 25 | Fe(NO3)3·9H2O | 96 ± 0 | 102 ± 2 |
| Mn2+ | 25 | Mn(NO3)2·4H2O | 95 ± 2 | 100 ± 1 |
| Cd2+ | 25 | Cd(NO3)2·4H2O | 99 ± 5 | — |
| Ni2+ | 25 | Ni(NO3)2·6H2O | 100 ± 4 | 103 ± 1 |
| Pb2+ | 25 | Pb(NO3)2·6H2O | — | 99 ± 2 |
| Cr3+ | 25 | Cr(NO3)3·9H2O | 100 ± 4 | 97 ± 2 |
| Cl− | 10000 |
KCl | 95 ± 4 | 97 ± 1 |
| SO42− | 5000 | Na2SO4 | 98 ± 3 | 96 ± 6 |
3.3. Analytical performance
The analytical features of the suggested method were evaluated under the optimum conditions. The cycle results show that the M-PhCP adsorbent is stable for up to 100 runs without a decrease in the recoveries of Pb(II) and Cd(II). The average ± standard deviations of the Pb(II) and Cd(II) recoveries after 100 extraction/elution cycles were found to be 100 ± 0 and 97 ± 1, respectively.The calibration equations were [A = 0.0023 + 0.0053C] for lead and [A = 0.0002 + 0.2216C] for cadmium where A is the absorbance and C is the analyte concentration in mg L−1 linear with correlation coefficients of 0.9980 and 0.9978, respectively. The detection limits of the analytes, defined as 3 times the signal/slope (slope of the calibration curve), were 2.7 and 1.1 μg L−1 for Pb(II) and Cd(II). The quantification limits, which were defined as 10 times the signal/slope (slope of the calibration curve), were 9.1 and 3.6 μg L−1 for Pb(II) and Cd(II). The relative standard deviations (RSD, %), evaluated using the results of the analysis of seven replicates containing 30 μg L−1 of Pb(II), and Cd(II), were 3.7 and 4.6, respectively. The preconcentration factor (PF = the ratio of the highest sample volume to the eluent volume) was 80. Consumptive index (CI) is another effective way to evaluate the analytical performance of a preconcentration system. CI was also determined by using the slope of the sample volume (SV) to the experimental preconcentration factor (CI = SV/PF). CI was found as 0.5 mL.
3.4. Analytical applications
To evaluate the accuracy of the developed method, the TMDA 64.2 environmental water and SPS-WW2 waste water certified reference materials were analyzed, and the analytical results with the standard deviations are shown in Table 3. It was found that there was no significant difference between the results obtained and the certified results.| Certified reference material | Analyte | Found, μg L−1 | Certified value, μg L−1 | Recovery, % |
|---|---|---|---|---|
| TMDA-64.2 fortified water | Pb | 280 ± 27 | 288 | 97 |
| Cd | 266 ± 6 | 266 | 100 | |
| SPS-WW2 waste water | Pb | 513 ± 1 | 500 | 103 |
| Cd | 103 ± 5 | 100 | 103 |
To further ensure the accuracy of the magnetic SPE method, the developed method was also applied to the analysis of four water samples corresponding to the CRMs and the analytical results along with the recoveries for the spiked waters are given in Table 4. As can be seen in Table 4, a good correlation exists between the added and recovered amounts of lead and cadmium ions. The results for the analysis of the certified reference materials and the addition–recovery studies show that the developed magnetic SPE method is reliable and independent from effects of the matrix for the determination of lead and cadmium in water samples.
| Sample | Added, μg L−1 | Pb(II) | Cd(II) | ||
|---|---|---|---|---|---|
| Found, μg L−1 | Recovery, % | Found, μg L−1 | Recovery, % | ||
| a BDL: below the detection limit.b Mean ± standard deviation. | |||||
| River water | 0 | 52 ± 6a | — | BDLb | |
| 40 | 93 ± 9 | 101 | 41 ± 0 | 103 | |
| Dam water | 0 | BDL | — | BDL | — |
| 133 | 133 ± 0 | 100 | 141 ± 0 | 106 | |
| Well water | 0 | 45 ± 0 | — | 45 ± 0 | — |
| 133 | 178 ± 0 | 100 | 178 ± 0 | 100 | |
| Waste water | 0 | 158 ± 31 | — | 18 ± 0 | — |
| 200 | 347 ± 9 | 97 | 214 ± 5 | 98 | |
4. Conclusion
In this study, a new magnetic phosphorus-containing polymeric sorbent was successfully used as a new sorbent in solid phase extraction for the simple, fast and efficient preconcentration of trace Pb and Cd from environmental water samples without matrix interferences. The new solid-phase extractor, which has good physical and chemical properties like durability, porosity and purity, and, most importantly, resistance to mineral acids, bases and organic solvents over a long time, was designed for lead and cadmium determination in water samples by FAAS.The most important advantages of the developed method compared with other preconcentration methods in the literature are: rapid collection of the analyte from the adsorbent by use of magnet elution, which eliminates the time-consuming column passing or filtration operation when compared with column and filtration operations, few adsorbent requirements and a low limit of detection for analytes. The analyses of certified reference materials and recovery tests show that the developed method can be used for the rapid preconcentration/separation of Pb(II) and Cd(II) in various water samples.
References
- M. Yaman and A. Sasmaz, Spectrosc. Spectral Anal., 2013, 33, 1 CAS.
- S. Vellaichamy and K. Palanivelu, J. Hazard. Mater., 2011, 185, 1131 CrossRef CAS PubMed.
- M. Tuzen and M. Soylak, Food Chem., 2007, 101, 1378 CrossRef CAS PubMed.
- A. Baran, E. Bicak, S. H. Baysal and S. Onal, Bioresour. Technol., 2006, 98, 661 CrossRef PubMed.
- M. Tuzen, A. Sari, D. Mendil and M. Soylak, J. Hazard. Mater., 2009, 169, 263 CrossRef CAS PubMed.
- M. Soylak, S. Saracoglu, U. Divrikli and L. Elci, Trace Elem. Electrolytes, 2001, 18, 47 CAS.
- F. Shah, E. Yilmaz, T. G. Kazi, H. I. Afridi and M. Soylak, Anal. Methods, 2012, 4, 4091 RSC.
- J. A. Baig, T. G. Kazi, A. Q. Shah, G. A. Kandhro, H. I. Afridi, M. B. Arain, M. K. Jamali and N. Jalbani, Ecotoxicol. Environ. Saf., 2010, 73, 914 CrossRef PubMed.
- P. Bulai, C. Balan, C. Cojocaru and M. Macoveanu, Environ. Eng. Manag. J., 2009, 8, 1413 CAS.
- S. Tajik and M. A. Taher, Desalination, 2011, 278, 57 CrossRef CAS PubMed.
- M. Ghaedi, H. Tavallali, A. Shokrollahi, M. Zahedi, M. Montazerozohori and M. Soylak, J. Hazard. Mater., 2009, 166, 1441 CrossRef CAS PubMed.
- Q. Li, Y. Chen, H. Fu, Z. Cui, L. Shi, L. Wang and Z. Liu, J. Hazard. Mater., 2012, 227–228, 148 CrossRef CAS PubMed.
- C. Duran, D. Ozdes, D. Sahin, V. N. Bulut, A. Gundogdu and M. Soylak, Microchem. J., 2011, 98, 317 CrossRef CAS PubMed.
- C. Karadas, Water, Air, Soil Pollut., 2014, 225, 2150 CrossRef.
- D. K. Acar and D. Kara, Water, Air, Soil Pollut., 2014, 225, 1864 CrossRef.
- M. Tuzen, I. Karaman, D. Citak and M. Soylak, Food Chem. Toxicol., 2009, 47, 1648 CrossRef CAS PubMed.
- E. Sahmetlioglu, E. Yilmaz, E. Aktas and M. Soylak, Talanta, 2014, 119, 447 CrossRef CAS PubMed.
- Y. B. Zhu, Bunseki Kagaku, 2007, 56, 895 CAS.
- M. Rajabi, S. Asemipour, B. Barfi, M. R. Jamali and M. Behzad, J. Mol. Liq., 2014, 194, 166 CrossRef CAS PubMed.
- I. López-García, Y. Vicente-Martínez and M. Hernández-Córdoba, Spectrochim. Acta, Part B, 2014, 101, 93 CrossRef PubMed.
- R. Lertlapwasin, N. Bhawawet, A. Imyim and S. Fuangswasdi, Sep. Purif. Technol., 2010, 72, 70 CrossRef CAS PubMed.
- M. Krawczyk, M. Jeszka-Skowron and H. Matusiewicz, Microchem. J., 2014, 117, 138 CrossRef CAS PubMed.
- J. Sun, Q. Liang, Q. Han, X. Zhang and M. Ding, Talanta, 2015, 132, 557 CrossRef CAS PubMed.
- M. Rezvani, A. A. Asgharinezhad, H. Ebrahimzadeh and N. Shekari, Microchim. Acta, 2014, 181, 1187 CrossRef.
- H. Parham and N. Rahbar, Talanta, 2009, 80, 664 CrossRef CAS PubMed.
- L. Chen, T. Wang and J. Tong, TrAC, Trends Anal. Chem., 2011, 30, 1095 CrossRef CAS PubMed.
- M. H. Mashhadizadeh and Z. Karami, J. Hazard. Mater., 2011, 190, 1023 CrossRef CAS PubMed.
- Y. Wang, X. Luo, J. Tang, X. Hua, Q. Xua and C. Yang, Anal. Chim. Acta, 2012, 713, 92 CrossRef CAS PubMed.
- I. S. Ibarra, J. A. Rodriguez, J. M. Miranda, M. Vega and E. Barrado, J. Chromatogr. A, 2011, 1218, 2196 CrossRef CAS PubMed.
- M. Soylak and E. Yilmaz, Desalination, 2011, 275, 297 CrossRef CAS PubMed.
- R. M. Alosmanov, Patent of Azerbaijan Republic, No: 0108, 2003.
- R. M. Alosmanov, A. A. Azizov and A. M. Maharramov, Russ. J. Gen. Chem., 2011, 81, 7 CrossRef.
- M. M. Rafi, K. S. Z. Ahmed, K. P. Nazeer, D. S. Kumar and M. Thamilselvan, Appl. Nanosci., 2015 DOI:10.1007/s13204-014-0344-z.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739 CrossRef CAS.
- L. O. dos Santos and V. A. Lemos, Water, Air, Soil Pollut., 2014, 225, 2086 CrossRef.
- F. A. Aydin and M. Soylak, Talanta, 2007, 72, 187 CrossRef CAS PubMed.
- J. G. Chen, H. W. Chen, S. H. Chen, L. Lin and Y. Y. Zhong, Chem. Res. Chin. Univ., 2007, 23, 143 CrossRef CAS.
- M. Soylak and L. Elci, J. Trace Microprobe Tech., 2000, 18, 397 CAS.
- M. Rezvani, H. Ebrahimzadeh, A. Aliakbari, A. Khalilzadeh, M. Kasaeian and M. M. Amini, J. Sep. Sci., 2014, 37, 2559 CrossRef CAS PubMed.
- A. Samadi and M. Amjadi, Microchim. Acta, 2015, 182, 257 CrossRef CAS.
- G. H. Mirzabe and A. R. Keshtkar, J. Radioanal. Nucl. Chem., 2015, 303, 561 CrossRef CAS.
- H. Tavallali, G. Deilamy-Rad and H. Ansari, Iran. J. Anal. Chem., 2014, 1, 87 Search PubMed.
- H. Shirkhanloo, A. Khaligh, H. Zavvar Mousavi and A. Rashidi, Int. J. Environ. Anal. Chem., 2015, 95, 16 CrossRef CAS.
- V. N. Bulut, M. Tufekci, C. Duran, M. Soylak and H. Kantekin, Clean, 2010, 38, 678 CAS.
- T. Li and J. Yang, J. Iran. Chem. Soc., 2015, 12, 367 CrossRef CAS.
- A. Gundogdu, D. Ozdes, C. Duran, V. N. Bulut, M. Soylak and H. B. Senturk, Chem. Eng. J., 2009, 153, 62 CrossRef CAS PubMed.
- H. Z. Mousavi and J. Derakhshankhah, J. AOAC Int., 2014, 97, 1707 CrossRef CAS PubMed.
- S. Baytak and A. R. Turker, Turk. J. Chem., 2004, 28, 243 CAS.
- M. Hossein, N. Dalali and A. Karimi, Turk. J. Chem., 2010, 34, 805 CAS.
- H. B. Senturk, A. Gundogdu, V. N. Bulut, C. Duran, M. Soylak, L. Elci and M. Tufekci, J. Hazard. Mater., 2007, 149, 317 CrossRef CAS PubMed.
- E. Yilmaz and M. Soylak, Talanta, 2013, 116, 882 CrossRef CAS PubMed.
- S. Saracoglu, M. Soylak and L. Elci, Can. J. Anal. Sci. Spectrosc., 2001, 46, 123 CAS.
| This journal is © The Royal Society of Chemistry 2015 |