
Influence of n-octanol in mobile phase on QSRRs of lipophilicity and retention mechanism of acidic and basic compounds in RP-HPLC
Shu-ying Hanab,
Chao Liangb,
Hui-min Yua,
Jun-qin Qiaob,
Xin Geb and
Hong-zhen Lian*b
aCollege of Pharmacy, Nanjing University of Chinese Medicine, 138 Xianlin Avenue, Nanjing 210023, China
bState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry & Chemical Engineering and Center of Materials Analysis, Nanjing University, 22 Hankou Road, Nanjing 210093, China. E-mail: hzlian@nju.edu.cn; Fax: +86-25-83325180; Tel: +86-25-83686075
First published on 17th March 2015
Abstract
The influence of n-octanol additive agent on the retention behavior, the uniformity of the retention mechanism, as well as the quantitative structure–retention relationships (QSRRs) of weak acidic and basic compounds on reversed-phase high performance liquid chromatography (RP-HPLC) was systematically discussed in this paper, especially for the QSRRs of logarithm of apparent n-octanol/water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K′′ow) and logarithm of retention factor extrapolated to neat aqueous mobile phase (logkw(o)), which have not been discussed in other studies to date. For this purpose, the aqueous fraction of mobile phase was saturated with n-octanol and 0.25% (v/v) n-octanol was added into organic modifier. Eleven substituted benzoic acids, as well as fifteen anilines or pyridines were selected to establish QSRR models by using different types of ion-suppressors. The results indicated that the roles of n-octanol were different in various systems. For acids compounds, if perchloric acid (strong acid) acts as an ion-pair agent, the silanophilic interaction between solutes and residual silanol groups of alkyl-silica stationary phase can be ignored, and n-octanol/water partition and chromatographic process are homo-energetic. In this case, n-octanol acts only as organic modifier. However, if acetic acid (weak acid) or phosphoric acid/potassium dihydrogen phosphate (buffer salt) were used as the ion-suppressor, n-octanol is not only an organic modifier, but also a masking agent of free silanols. For weak bases, if ammonium chloride–ammonia was employed as the ion-suppressor, the addition of n-octanol will make QSRRs correlation significantly worse. Therefore, for studying on QSRRs of lipophilicity and retention behavior of acidic and basic compounds, n-octanol is only recommended for acidic system, and strong monoprotic acids, e.g., perchloric acid, are recommended as the ion-suppressors.
K′′ow) and logarithm of retention factor extrapolated to neat aqueous mobile phase (logkw(o)), which have not been discussed in other studies to date. For this purpose, the aqueous fraction of mobile phase was saturated with n-octanol and 0.25% (v/v) n-octanol was added into organic modifier. Eleven substituted benzoic acids, as well as fifteen anilines or pyridines were selected to establish QSRR models by using different types of ion-suppressors. The results indicated that the roles of n-octanol were different in various systems. For acids compounds, if perchloric acid (strong acid) acts as an ion-pair agent, the silanophilic interaction between solutes and residual silanol groups of alkyl-silica stationary phase can be ignored, and n-octanol/water partition and chromatographic process are homo-energetic. In this case, n-octanol acts only as organic modifier. However, if acetic acid (weak acid) or phosphoric acid/potassium dihydrogen phosphate (buffer salt) were used as the ion-suppressor, n-octanol is not only an organic modifier, but also a masking agent of free silanols. For weak bases, if ammonium chloride–ammonia was employed as the ion-suppressor, the addition of n-octanol will make QSRRs correlation significantly worse. Therefore, for studying on QSRRs of lipophilicity and retention behavior of acidic and basic compounds, n-octanol is only recommended for acidic system, and strong monoprotic acids, e.g., perchloric acid, are recommended as the ion-suppressors.
1. Introduction
Lipophilicity, generally expressed by n-octanol/water partition coefficient (Kow), constitutes an important physicochemical parameter conventionally used in quantitative structure–retention relationships (QSRRs) for various bioactive compounds including pharmaceuticals and natural products.1–5 A variety of experimental methods are available to measure Kow. The shake-flask method (SFM) and slow-stirring method (SSM) are often regarded as classical, but in practice they are tedious and time consuming.6 The reversed-phase high performance liquid chromatography (RP-HPLC) is a promising alternative to SFM/SSM, having such advantages as high throughput, insensitivity to impurities and broad lipophilicity range. In the RP-HPLC method, Kow is derived from kw, that is the retention factor (k) value extrapolated to neat aqueous mobile phase to eliminated organic solvent effect. This straight relationship between logKow and logkw is also known as Collander equation.7,8 It has been demonstrated the usefulness of logkw when investigating series of solutes covering a broad lipophilicity range.9,10 In general, Collander equation is limited to neutral solutes. In fact, most environmental and biomedical molecules are more or less dissociated, therefore, buffers including acids and bases are added into the mobile phases to suppress the dissociation of compounds with acid–base properties, which results in improved chromatographic retention and peak shape in RP-HPLC.11–13 However, for ionizable acidic or basic compounds, the dissociation is completely suppressed only when the pH of mobile phase was adjusted to at least 2 pH units lower or higher than pKa of the solute, which means that very strong acidity or alkalinity of mobile phase is required for those compounds with extreme pKa values, decreasing the life of chromatographic columns as well as apparatus. The apparent n-octanol/water partition coefficient (K′′ow) has been proposed to correct Kow so as to describe the lipophilicity of ionizable solutes more precisely. A better linear relationship relating logK′′ow with logkw than that relating logKow with logkw has been revealed and applied successfully to Kow measurement as well as retention behavior prediction of weak acids or bases in our previous studies.14–18
Octadecyl silica (ODS) is the most widely used packing material for reversed-phase stationary phase that is appropriate for lipophilicity determination by RP-HPLC. The linear correlations between logKow (logK′′ow) and logkw are satisfactory for structurally related solutes. However, the interference of silanophilic interaction between investigated compounds and residual silanol groups might lead to inferior linearity, especially in the case of ionizable solutes.19,20 An approach explored to reduce silanol activity was the utilization of new stationary phases possessing functional groups introduced to eliminate free silanols, such as end-capped base deactivated silica (BDS), LC-ABZ, Discovery-RP-Amide-C16, and polymer-based octadecylpolyvinyl (ODP).21–24 Although many works have demonstrated that the logKow(logK′′ow) − logkw correlations were better with these new stationary phases, it has been reported that the retention mechanism on these new stationary phases and n-octanol/water partitioning might be controlled by a different equilibrium of structural properties. Thus, the resulting data are probably not suitable to represent the classical Kow values.25 Moreover, the problems associated with long-term stability of these stationary phases should be considered, especially in the long period experiment. Silanophilic interaction can also be strongly reduced or even suppressed by the addition of a masking agent (e.g., n-decylamine, triethylamine, and n-octanol, etc.) into the mobile phases.26 The masking agent-coated stationary phases provide a more realistic model for the n-octanol/water partition system than widely used chemically bonded stationary phases, and moreover, supply a more flexible operation and lower cost than new stationary phases. However, n-decylamine and triethylamine cannot be applied for acidic solutes.27 Instead, the employment of n-octanol in mobile phase has been reported in many works,28–30 but the effect of n-octanol is still quite unclear.
In this present study, the influence of n-octanol on logK′′ow − logkw QSRR models for both weak acidic and basic compounds was examined in respect to the final retention outcome as appropriate measure for n-octanol/water partitioning simulation. The QSRRs of logK′′ow − logkw modeling by 11 substituted benzoic acids using three types of ion-suppressors, i.e., strong acid, weak acid and buffer salt, respectively, as well as by 15 anilines or pyridines by using a kind of buffer salt were compared. The influence of n-octanol on the uniformity of the retention mechanism was also investigated. Furthermore, the relationships of retention behaviors in presence and absence of n-octanol were studied at different elution conditions, and these retention factors were correlated with logK′′ow values, in an attempt to establish and compare relevant models for ionizable compounds.
2. Experimental
2.1 Chemicals
Water for mobile phase was Wahaha purified water (Wahaha Group, Hangzhou, China). The mobile phases were prepared from methanol (HPLC grade, Merck, Darmstadt, Germany) and aqueous acidic–basic solution. The employed ion-suppressors are acetic acid (analytical-reagent grade, Sinopharm Group Chemical Reagent, Shanghai, China), perchloric acid (70–72%, analytical-reagent grade, Nanjing Chemical Reagent, Nanjing, China), phosphoric acid (≥85%, guaranteed-reagent grade, Sinopharm Group Chemical Reagent), potassium dihydrogen phosphate (≥99.5%, analytical-reagent grade, Nanjing Chemical Reagent), ammonium chloride (>99%, analytical-reagent grade, Shanghai Inorganic Chemical Industry Research Institute, Shanghai, China), and ammonia solution (25%, analytical-reagent grade, Nanjing Chemical Reagent). n-Octanol (99%) was purchased from Acros Organics (New Jersey, USA). Table 1 lists all substances investigated in this experiment with their reliable literature logKow and pKa data. They were all with the purity of 98% or higher, checked by RP-HPLC, and then used without further purification. Stock solutions of these compounds were respectively prepared in methanol (ca. 1.0 mg mL−1) and stored in refrigerator before use.
| Compounds | LogKowa |
pKab | LogK′′ow |
||
|---|---|---|---|---|---|
| Mobile phase pH | |||||
| a Only reliable SFM/SSM data were adopted: a1,a3,a4,a8,a9,a11,a12,a15,a16,a17,a18,a20,a22,a26,a27 from ref. 31; a2,a7,a10 from ref. 32; a5,a6 from ref. 33; a13 from ref. 34; a14,a23,b23 from ref. 35; a19 from ref. 36; a21 from ref. 37; a24 from ref. 38; a25 from ref. 39.b b1–b11 From ref. 40; b12,b15 from ref. 41; b13,b17,b24 from ref. 42; b14,b22 from ref. 43; b16,b18 from ref. 44; b19,b27 from ref. 45; b21,b26 from ref. 46; b20,b25 from ref. 47. | |||||
| Weak acids | 2.80 | 3.20 | 3.60 | ||
| Benzoic acid | 1.87a1 | 4.20b1 | 1.85 | 1.83 | 1.77 |
| 2-Methylbenzoic acid | 2.18a2 | 3.90b2 | 2.15 | 2.10 | 2.00 |
| 3-Methylbenzoic acid | 2.37a3 | 4.27b3 | 2.36 | 2.33 | 2.28 |
| 4-Methylbenzoic acid | 2.27a4 | 4.36b4 | 2.26 | 2.24 | 2.20 |
| 4-Ethylbenzoic acid | 2.89a5 | 4.35b5 | 2.88 | 2.86 | 2.82 |
| 4-(1-Methylethyl)benzoic acid | 3.40a6 | 4.35b6 | 3.27 | 3.25 | 3.21 |
| 2-Chlorobenzoic acid | 2.05a7 | 2.88b7 | 1.79 | 1.56 | 1.25 |
| 3-Chlorobenzoic acid | 2.68a8 | 3.83b8 | 2.64 | 2.59 | 2.48 |
| 4-Chlorobenzoic acid | 2.65a9 | 3.99b9 | 2.62 | 2.58 | 2.50 |
| 2-Bromobenzoic acid | 2.20a10 | 2.85b10 | 1.92 | 1.69 | 1.38 |
| 3-Bromobenzoic acid | 2.87a11 | 3.81b11 | 2.83 | 2.77 | 2.66 |
|
|||||
| Weak bases | 7.40 | 9.00 | |||
| 4-Fluoroaniline | 1.15a12 | 4.65b12 | 1.15 | 1.15 | |
| N,N-Diethylaniline | 3.31a13 | 6.61b13 | 3.24 | 3.31 | |
| 2,4,6-Trimethylpyridine | 1.88a14 | 7.25b14 | 1.65 | 1.87 | |
| 2-Methylaniline | 1.32a15 | 4.45b15 | 1.32 | 1.32 | |
| N,N-Dimethylaniline | 2.31a16 | 5.07b16 | 2.31 | 2.31 | |
| 2-Methoxyaniline | 1.18a17 | 4.49b17 | 1.18 | 1.18 | |
| 4-Iodoaniline | 2.34a18 | 3.78b18 | 2.34 | 2.34 | |
| 2-Methylpyridine | 1.11a19 | 5.97b19 | 1.09 | 1.11 | |
| N,N-Dimethylbenzylamine | 1.98a20 | 8.80b20 | 0.56 | 1.77 | |
| 2,6-Dimethylpyridine | 1.68a21 | 6.72b21 | 1.60 | 1.68 | |
| 3-Methylaniline | 1.40 a22 | 4.73b22 | 1.40 | 1.40 | |
| 2-Ethylpyridine | 1.69a23 | 5.97b23 | 1.67 | 1.69 | |
| 4-Ethoxyaniline | 1.24a24 | 5.20b24 | 1.24 | 1.24 | |
| 4-Isopropylphenylamine | 2.23a25 | 4.85b25 | 2.23 | 2.23 | |
| 2-Amino-4-methylpyridine | 0.56a26 | 7.38b26 | 0.27 | 0.55 | |
| Benzidine | 1.34a27 | 3.85b27 | 0.34 | 1.34 | |
2.2 Apparatus
A Waters 2695 Alliance separation module (Milford, MA, USA) was employed consisting of a vacuum degasser, a quaternary pump and an auto-sampler, and a Waters 996 photodiode-array (PDA) detector set at the respective optimum absorption wavelength for each eluted compound. The chromatographic column used was an Agela Venusil XBP C18, 5 μm, 150 mm × 2.1 mm i.d. (Bonna-Agela Technologies, Tianjin, China) maintained at 30 °C. Data acquisition and processing were performed on a Waters Empower chromatography manager system. All experimental retention times (tR) were obtained by averaging the results of at least three independent injections at 0.2 mL min−1 mobile phase flow rate.The pH values of mobile phase were measured with a SevenMulti electrochemical analytical meter (Metter-Toledo Instrum., Schwerzenbach, Switzerland). All pH readings were carried out in wwpH scale.48
2.3 Procedure
For weak acids: solutes were eluted by the mobile phase consisting of methanol and water at pH 2.80, 3.20 and 3.60. Each mobile phase pH was adjusted by acetic acid (AA), perchloric acids (PA), and 20 mM phosphoric acid/potassium dihydrogen phosphate (PBS), respectively.For weak bases: solutes were eluted by the mobile phase consisting of methanol and water at pH 7.40 and 9.00. Both mobile phase pH were adjusted by ammonium chloride–ammonia buffer solution.
Two sets of measurements were conducted for all compounds. In one set, a 0.25% (v/v) amount of n-octanol was added to methanol, and n-octanol saturated water was used to prepare the aqueous fraction of mobile phase, which is symbolized as subscript (o) in the text and tables. In another set, the mobile phase condition was the same as that used in the first set except for absence of n-octanol in the eluents. At each pH adjusted by every ion-suppressor, at least four different methanol contents (φ) were required to elute each solute according to its lipophilicity. The tR value was recorded at each methanol–aqueous solution ratio, then corrected by dual-point retention time correction (DP-RTC) using 2-chlorobenzoic acid and 3-bromobenzoic acid (for acidic compounds), and 4-fluoro aniline, N,N′-diethyl aniline, and 2,4,6-trimethyl pyridine (for basic compounds) as “anchor compounds”. The k value was calculated according to the equation k = (tR − t0)/t0, where t0 was determined by using sodium nitrate eluted on the “standard column”. The detailed process of DP-RTC refers to our previous work.49 For each solute, the logarithm of k was plotted against φ, and logkw of the solute was subsequently obtained by extrapolation of k to neat aqueous mobile phase via Snyder-Soczewinski equation.7 The literature Kow value of each compound was calibrated to the corresponding K′′ow.14–18 Then the correlations relating logK′′ow and logkw of investigated compounds at various elution conditions were derived with different ion-suppressors at different pH.
The statistical analysis for regression model was accomplished by SPSS V16.0.0 (SPSS, Chicago, Illinois, USA) and MATLAB Software V7.10.0 (R2010.a) (The MathWorks, Natick, MA, USA).
3. Results and discussion
3.1 Effect of n-octanol on the retention mechanism of acidic–basic solutes in different buffering systems
It was observed in the experiment that both logkw(o) and logkw of acidic solutes decreased as mobile phase pH increased because of the dissociation of these solutes at high pH; in the same way, for basic solutes, both logkw(o) and logkw increased as mobile phase pH increased. For a given elution condition, addition of hydrophobic n-octanol would strengthen the eluting power of mobile phase, thereupon weaken the retention of solutes regardless of ion-suppressor or mobile phase pH used, i.e., logkw(o) < logkw, which indicated that n-octanol firstly acted as organic modifier in this work. Table 2 and Fig. 1 show the relationships between logkw(o) and logkw of investigated compounds, respectively, using different ion-suppressors at various mobile phase pH.
kw(o) and logkw of investigated compounds for various ion-suppressors at different pH (95% confidence limits are in parentheses)
| Ion-suppressors | pH | Logkw(o) − logkw |
|||
|---|---|---|---|---|---|
| Slope | Intercept | R2 | Rcv2 | ||
| Weak acids | |||||
| Perchloric acid | 2.80 | 0.97 (0.03) | −0.32 (0.06) | 0.993 | 0.989 |
| 3.20 | 0.96 (0.03) | −0.35 (0.06) | 0.991 | 0.987 | |
| 3.60 | 0.99 (0.02) | −0.40 (0.05) | 0.994 | 0.992 | |
| Acetic acid | 2.80 | 1.00 (0.03) | −0.40 (0.06) | 0.992 | 0.987 |
| 3.20 | 1.01 (0.02) | −0.44 (0.05) | 0.995 | 0.993 | |
| 3.60 | 1.11 (0.06) | −0.58 (0.14) | 0.983 | 0.943 | |
| PBS | 2.80 | 0.91 (0.02) | −0.16 (0.05) | 0.994 | 0.991 |
| 3.20 | 1.03 (0.04) | −0.51 (0.08) | 0.987 | 0.978 | |
| 3.60 | 1.17 (0.05) | −0.82 (0.11) | 0.982 | 0.970 | |
|
|||||
| Weak bases | |||||
| Ammonium chloride-ammonia | 7.40 | 1.01 (0.06) | −0.41 (0.09) | 0.951 | 0.947 |
| 9.00 | 1.12 (0.09) | −0.67 (0.15) | 0.916 | 0.902 | |
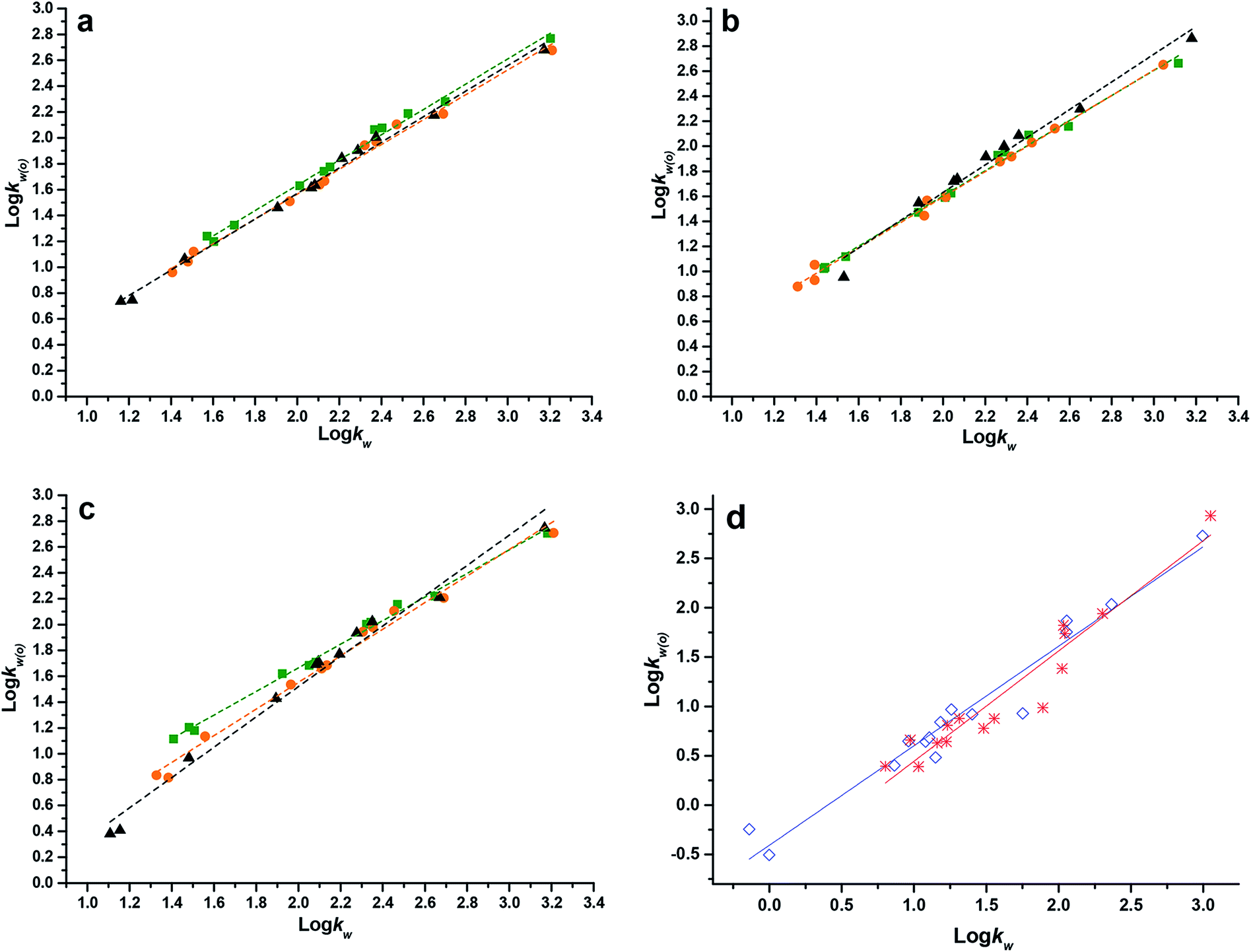 | ||
Fig. 1 Comparison of relationships between logkw(o) and logkw of investigated acidic and basic compounds at different mobile phase pH adjusted by perchloric acid (a); acetic acid (b); PBS (c); and ammonium chloride–ammonia (d). pH:  2.80; 2.80;  3.20; 3.20;  3.60; 3.60;  7.40; and 7.40; and  9.00. 9.00. | ||
For acidic compounds, as shown in Fig. 1a, the slopes of fitting lines obtained by using perchloric acid as the ion-suppressor at different pH were significantly consistent. When mobile phase pH was adjusted by acetic acid (Fig. 1b), the slope obtained at pH 3.60 showed a distinct difference in comparison with others obtained at lower pH. The discrepancy of these slopes acquired at various pH even increased by using PBS as the ion-suppressor, which can be seen in Fig. 1c. The reason for high consistency of slopes of fitting lines at different pH adjusted by perchloric acid is that perchloric acid can be considered as an ion-pair agent, prevailing over investigated acidic solutes to occupy residual silanol sites on C18 stationary phase, thereupon the solutes would not interact with these residual silanols. Hence, as listed in Table 2, variation of mobile phase pH had little effect on fitting equations. The only slightly difference in intercepts of fitting lines was that the retention times of acidic solutes are influenced to various degrees by additional organic modifier n-octanol at different pH. In contrast, the change in slopes of fitting lines obtained by using acetic acid or PBS as the ion-suppressor at higher pH shown in Table 2 and Fig. 1 was probably due to the role of “masking-agent” played by n-octanol. As we know, the proportion of dissociated acidic solutes increases with pH increases, thus the high affinity between anions and residual silanol of reversed-phase C18 cannot be neglected in high mobile phase pH. This affinity inevitably prolonged retention of acidic solutes, especially for hydrophilic ones (logkw < 2.0). As a result, retention times of these solutes dropped slower at high pH. However, these residual silanol groups may be covered by n-octanol added into mobile phase, which makes the affinity weaker, thereby the decrease in retention of solutes would not slow down. Consequently, this variation in retention mechanism caused by n-octanol resulted in a noticeable deviation of fitting slopes obtained at high pH from the ones at lower pH. In view of this, it is indicated that n-octanol is not only an organic modifier, but also a masking-agent of silanol when weak acids or buffers are used as ion-suppressors.
For basic compounds, as shown in Fig. 1d and Table 2, the tendency of fitting lines obtained by using ammonium chloride–ammonia as the ion-suppressor at different pH was just similar with the ones obtained by using PBS for acidic compounds, which means the n-octanol had the analogous effect in acidic or alkaline system when buffers were used as ion-suppressors.
3.2 Effect of n-octanol on the retention behavior of acidic solutes by using different ion-suppressors
The retention factors of investigated acidic compounds extrapolated back to 100% aqueous solution at the same elution condition (except by using three different ion-suppressors) were approximate, but still had a little discrepancy. As shown in Fig. 2a and c, no matter whether n-octanol was added into mobile phase or not, logkw of each solute at lower pH (2.80) got its largest value when perchloric acid was used as the ion-suppressor, and got the least value when acetic acid was used as the ion-suppressor, that is, logkw,PA > logkw,PBS > logkw,AA, and logkw(o),PA > logkw(o),PBS > logkw(o),AA. The reason for shortest retention of solutes in acetic acid containing mobile phase at low pH was due to the role of organic modifier played by non-dissociated acetic acid, which had been explained detailed in our previous work.14–16 Moreover, it can be seen that retention of solutes obtained by using acetic acid as the ion-suppressor became even shorter when n-octanol was present in mobile phase. The possible reason is that n-octanol blended with acetic acid as the mixed organic modifier, which further enhanced eluting power of the mobile phase. However, this phenomenon changed when pH increased. The well overlapped plots shown in Fig. 2b indicated that without n-octanol in mobile phase, logkw values obtained by using different ion-suppressors had high consistency at pH 3.60, namely, logkw,PA ≈ logkw,PBS ≈ logkw,AA. For acetic acid, the neutral form of acetic acid was decreased markedly when pH increased, thus its role of organic modifier was negligible. In contrast, the affinity between anions and residual silanol groups mentioned above increased the retention of acidic solutes at high pH. The similar affinity also existed when PBS was used as the ion-suppressor. That is to say, logkw,PA had greater decrease than logkw,AA and logkw,PBS did. Thus, the difference in retention factors of acidic solutes at three different ion-suppressors was eliminated in this situation. However, Fig. 2d illustrates that if n-octanol was added into mobile phase, the largest logkw value of the solute was obtained by using acetic acid as the ion-suppressor, and logkw(o),PBS was close to logkw(o),PA for each solute, i.e., logkw(o),AA > logkw(o),PA ≈ logkw(o),PBS. Although use of n-octanol decreased retention times of all the solutes by using various ion-suppressors, it seems that logkw(o),PA decreased slightly larger than other two. Just as discussed above, the decrease in logkw(o),PA was attributed to the enhanced eluting power of mobile phase by adding hydrophobic n-octanol. However, for acetic acid and PBS, a majority of n-octanol was used as a masking-agent to cover residual silanol, so the retention decrease caused by n-octanol as organic modifier was minor.
 | ||
Fig. 2 Comparison of extrapolated retention factor of investigated acidic compounds at different mobile phase pH adjusted by various ion-suppressors. (a) pH 2.80 without n-octanol in mobile phase; (b) pH 3.60 without n-octanol in mobile phase; (c) pH 2.80 with n-octanol in mobile phase; and (d) pH 3.60 with n-octanol in mobile phase. Ion-suppressor:  Perchloric acid; Perchloric acid;  Acetic acid; and Acetic acid; and  PBS. PBS. | ||
3.3 Effect of n-octanol on the QSRRs of lipophilicity in different buffering systems
Table 3 lists relationships between logK′′ow(logKow) and logkw(logkw(o)) for 11 substituted benzoic acids, as well as for 15 anilines or pyridines eluted by various methanol–aqueous solutions at different mobile phase pH. Just the same as we concluded in previous work, better linearity between logK′′ow and logkw(logkw(o)) than that between logKow and logkw(logkw(o)) was obtained under all the elution conditions, which revealed that no matter whether n-octanol was added into mobile phase, K′′ow always should be used to describe lipophilicity of ionizable solutes. In addition, high consistency of logK′′ow − logkw(logkw(o)) linear fittings was observed at different pH adjusted by the same ion-suppressor in most cases, although corresponding logKow − logkw(logkw(o)) equations varied. When perchloric acid was used as the ion-suppressor, slopes of logK′′ow − logkw(logkw(o)) correlations were all close to 1, implying the apparent n-octanol/water partitioning and chromatographic retention are homo-energetic processes, which means logkw or logkw(o) both can simulate logK′′ow well. Moreover, intercepts of logK′′ow − logkw and logK′′ow − logkw(o) correlations established at different pH were invariable, respectively. The intercepts of logK′′ow − logkw(o) were larger than that of logK′′ow − logkw, which may be due to the effect of organic modifier performed by n-octanol. When acetic acid or PBS was used as the ion-suppressor, slopes of logK′′ow − logkw(logkw(o)) correlations were also all close to 1 with the exception of slopes of logK′′ow − logkw(o) obtained at pH 3.60. The negative deviation of the slopes turned out that the apparent n-octanol/water partitioning and chromatographic retention are not homo-energetic processes.
K′′ow (logKow) and logkw(logkw(o)) for 11 substituted benzoic acids and 15 weak bases eluted by various methanol–aqueous buffer solutions at different mobile phase pH (95% confidence limits are in parentheses)
| Without n-octanol | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH | Ion-suppressor | LogKow − logkw |
LogK′′ow − logkw |
||||||||||
| Slope | Intercept | R2 | Rcv2 | S.D. | F | Slope | Intercept | R2 | Rcv2 | S.D. | F | ||
| 2.80 | Perchloric acid | 0.88 (0.07) | 0.54 (0.15) | 0.946 | 0.931 | 0.10 | 176.75 | 0.99 (0.04) | 0.22 (0.09) | 0.985 | 0.982 | 0.06 | 676.32 |
| Acetic acid | 0.84 (0.07) | 0.73 (0.15) | 0.937 | 0.918 | 0.11 | 150.95 | 0.96 (0.04) | 0.42 (0.08) | 0.986 | 0.983 | 0.06 | 681.27 | |
| PBS | 0.80 (0.07) | 0.80 (0.16) | 0.921 | 0.890 | 0.13 | 118.05 | 0.91 (0.04) | 0.48 (0.08) | 0.985 | 0.983 | 0.06 | 672.76 | |
| 3.20 | Perchloric acid | 0.77 (0.08) | 0.83 (0.18) | 0.897 | 0.855 | 0.14 | 87.81 | 0.99 (0.04) | 0.21 (0.09) | 0.984 | 0.978 | 0.07 | 609.09 |
| Acetic acid | 0.79 (0.08) | 0.87 (0.17) | 0.905 | 0.860 | 0.14 | 95.76 | 1.01 (0.04) | 0.29 (0.08) | 0.985 | 0.979 | 0.07 | 655.43 | |
| PBS | 0.74 (0.09) | 0.92 (0.21) | 0.857 | 0.855 | 0.17 | 60.77 | 0.97 (0.04) | 0.29 (0.08) | 0.986 | 0.983 | 0.07 | 705.83 | |
| 3.60 | Perchloric acid | 0.68 (0.10) | 1.10 (0.21) | 0.820 | 0.736 | 0.19 | 46.68 | 1.02 (0.04) | 0.14 (0.08) | 0.984 | 0.978 | 0.08 | 622.64 |
| Acetic acid | 0.70 (0.10) | 1.06 (0.22) | 0.822 | 0.739 | 0.19 | 47.10 | 1.04 (0.04) | 0.10 (0.09) | 0.985 | 0.977 | 0.08 | 664.29 | |
| PBS | 0.65 (0.10) | 1.16 (0.22) | 0.791 | 0.690 | 0.21 | 38.84 | 0.99 (0.04) | 0.21 (0.08) | 0.985 | 0.981 | 0.08 | 668.15 | |
| 7.40 | Ammonium chloride-ammonia | 0.66 (0.14) | 0.81 (0.21) | 0.621 | 0.602 | 0.42 | 23.98 | 0.89 (0.05) | 0.36 (0.08) | 0.951 | 0.942 | 0.17 | 270.13 |
| 9.00 | Ammonium chloride-ammonia | 1.09 (0.06) | −0.06 (0.11) | 0.941 | 0.941 | 0.16 | 227.00 | 1.08 (0.07) | −0.06 (0.12) | 0.952 | 227.00 | 0.15 | 278.57 |
| With n-octanol | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH | Ion-suppressor | LogKow − logkw(o) |
LogK′′ow − logkw(o) |
||||||||||
| Slope | Intercept | R2 | Rcv2 | S.D. | F | Slope | Intercept | R2 | Rcv2 | S.D. | F | ||
| 2.80 | Perchloric acid | 0.90 (0.07) | 0.84 (0.13) | 0.943 | 0.917 | 0.11 | 165.81 | 1.02 (0.03) | 0.55 (0.05) | 0.993 | 0.991 | 0.04 | 1428.35 |
| Acetic acid | 0.84 (0.07) | 1.07 (0.12) | 0.940 | 0.912 | 0.11 | 158.00 | 0.95 (0.03) | 0.81 (0.05) | 0.991 | 0.988 | 0.05 | 1141.40 | |
| PBS | 0.87 (0.08) | 0.95 (0.15) | 0.915 | 0.873 | 0.13 | 108.34 | 1.00 (0.04) | 0.65 (0.07) | 0.986 | 0.981 | 0.06 | 687.60 | |
| 3.20 | Perchloric acid | 0.80 (0.09) | 1.12 (0.15) | 0.896 | 0.841 | 0.14 | 87.31 | 1.03 (0.02) | 0.59 (0.04) | 0.995 | 0.992 | 0.04 | 1814.50 |
| Acetic acid | 0.77 (0.09) | 1.22 (0.15) | 0.889 | 0.830 | 0.15 | 80.95 | 1.00 (0.02) | 0.71 (0.04) | 0.994 | 0.992 | 0.04 | 1688.70 | |
| PBS | 0.70 (0.10) | 1.31 (0.18) | 0.820 | 0.711 | 0.19 | 46.56 | 0.92 (0.04) | 0.79 (0.07) | 0.982 | 0.971 | 0.07 | 534.12 | |
| 3.60 | Perchloric acid | 0.68 (0.10) | 1.38 (0.17) | 0.816 | 0.713 | 0.19 | 45.49 | 1.03 (0.03) | 0.57 (0.05) | 0.992 | 0.988 | 0.06 | 1191.33 |
| Acetic acid | 0.58 (0.11) | 1.53 (0.21) | 0.728 | 0.348 | 0.23 | 25.13 | 0.80 (0.04) | 0.93 (0.09) | 0.970 | 0.951 | 0.10 | 290.52 | |
| PBS | 0.53 (0.10) | 1.66 (0.18) | 0.713 | 0.553 | 0.24 | 25.83 | 0.84 (0.04) | 0.91 (0.07) | 0.979 | 0.971 | 0.09 | 476.14 | |
| 7.40 | Ammonium chloride-ammonia | 0.62 (0.14) | 1.11 (0.17) | 0.573 | 0.323 | 0.44 | 19.79 | 0.84 (0.06) | 0.75 (0.08) | 0.923 | 0.911 | 0.21 | 170.29 |
| 9.00 | Ammonium chloride-ammonia | 0.91 (0.08) | 0.67 (0.11) | 0.896 | 0.877 | 0.22 | 121.59 | 0.92 (0.07) | 0.65 (0.10) | 0.916 | 0.901 | 0.20 | 153.80 |
On the other hand, the statistical results summarized in Table 3 suggested that use of n-octanol did not improve the classical linearity between logKow and logarithm of extrapolated retention factor of investigated solutes. However, the improved linearity of logK′′ow − logkw(o) obtained by adding n-octanol into mobile phase was observed when perchloric acid was used as the ion-suppressor, indicating that use of n-octanol may reduce the difference of structure among investigated solutes, which further illustrated that K′′ow is the very parameter reflecting the real hydrophobicity of the ionizable solute in this buffering system. The linearity obtained with adding n-octanol was a little inferior at pH 3.60 when acetic acid or PBS was used as the ion-suppressor, confirming that there were some secondary interaction between n-octanol and weak acidic ion-suppressors, which led to the inconsistent mechanism between n-octanol/water partitioning and chromatographic retention. However, the linearity was still acceptable, indicating that these two processes are homeo-energetic. The statistical results summarized in Table 3 indicated that it is most suitable by using strong acid as the ion-suppressor for modeling logK′′ow − logkw(logkw(o)) correlations for weakly acidic compounds. Whereas, the linearity of logK′′ow − logkw relationship is inevitably affected by the secondary interaction when weak acid or buffer salt is used as the ion-suppressor.
However, the relationships between logK′′ow(logKow) and logkw(logkw(o)) for 15 basic compounds at different mobile phase pH listed in Table 3 indicated the disparate results. When n-octanol was added into the mobile phase, both of the linearity between logK′′ow(logKow) and logarithm of extrapolated retention factor of investigated bases turned worse, although better linearity between logK′′ow and logkw(logkw(o)) was still obtained at both pH in this work. The results indicated that n-octanol is not an appropriate additive agent for basic compounds in QSRRs.
4. Conclusions
In practice RP-HPLC, n-octanol is usually added into mobile phase to improve the QSRRs of lipophilicity and retention behavior of weakly ionizable compounds. The effect of n-octanol was mentioned in some works, but still has not a clear explanation. In this work, we discussed the influence of n-octanol on retention behavior and the related QSRR models of weak acids and bases. Three different types of ion-suppressor, i.e., strong acid, weak acid and buffer salt were used to adjust mobile phase pH for acidic solutes, and a buffer salt was used for basic ones. QSRRs of logK′′ow and logkw (logkw(o)) obtained by using different ion-suppressors were compared in detail. The logK′′ow − logkw(logkw(o)) correlations obtained by using perchloric acid as the ion-suppressor were in all cases linear with essentially unit slope, which means in this case the widely different retention behavior of the analytes on alkyl-silica stationary phases became homo-energetic. The dramatic homo-energetic effect of perchloric acid is believed to arise from its strong binding to the silanol groups as the ion-pair agent at the surface of the stationary phase. As a result, silanophilic interactions are so attenuated that retention occurs via solvophobic interactions only. In this case, n-octanol acted only as an organic modifier. However, the slopes of logK′′ow − logkw(o) correlations obtained by using acetic acid or phosphoric acid/potassium dihydrogen phosphate as the ion-suppressor deviated from 1 at high pH, indicating that in this situation n-octanol not only acted as an organic modifier, but also played a role as a masking agent of free silanol groups. In contrary, the logK′′ow −logkw(o) correlations obtained from weak bases by using ammonium chloride–ammonia as the ion-suppressor lead to an inferior result. In summary, it is suggested that for studying on QSRRs of lipophilicity and retention of acidic compounds on RP-HPLC, the usage of strong monoprotic acid, e.g. perchloric acid as the ion-suppressor, and n-octanol as the additive agent are recommended.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81303311, 21275069), National Basic Research Program of China (973 program, 2011CB911003), Natural Science Foundation of Jiangsu (BK20130958), Natural Science Foundation for Colleges of Jiangsu (13KJB150030), Priority Academic Program Development of Jiangsu Higher Education Institutions, Natural Science Foundation of Nanjing University of Chinese Medicine (12XZR27), National Science Funds for Creative Research Groups (21121091), and Analysis & Test Fund of Nanjing University.References
- H. Van de Waterbeemd and B. Testa, in Advances in Drug Research, ed. B. Testa, Academic Press, New York, 1987 Search PubMed.
- C. Hansch, A. J. Leo and D. Hoekman, Fundamentals and Applications in Chemistry and Biology in Exploring QSAR, American Chemical Society, Washington DC, 1995 Search PubMed.
- D. A. Smith, B. C. Jones and D. K. Walker, Med. Res. Rev., 1996, 16, 243–266 CrossRef CAS.
- H. Van de Waterbeemd, D. A. Smith, K. Beaumont and D. K. Walker, J. Med. Chem., 2001, 44, 1313–1333 CrossRef CAS.
- T. I. Oprea, J. Comput.-Aided Mol. Des., 2002, 16, 325–334 CrossRef CAS.
- A. Finizio, M. Vighi and D. Sandroni, Chemosphere, 1997, 34, 131–161 CrossRef CAS.
- L. R. Snyder, J. W. Dolan and J. R. Gant, J. Chromatogr. A, 1979, 165, 3–30 CrossRef CAS.
- T. Braumann, J. Chromatogr. A, 1986, 373, 191–225 CrossRef CAS.
- J. E. Garst and W. C. Wilson, J. Pharm. Sci., 1984, 73, 16l6–1629 Search PubMed.
- OECD, Guideline for Testing of Chemicals, No.117: Partition Coefficient (n-octanol/water)-High Performance Liquid Chromatography Method, 1989.
- R. E. A. Escott, P. G. McDowell and N. P. Porter, J. Chromatogr. A, 1991, 554, 281–292 CrossRef CAS.
- C. Crescenzi, A. Di Corcia, S. Marchese and R. Samperi, Anal. Chem., 1995, 67, 1968–1975 CrossRef CAS.
- R. B. Cole, Electrospray Ionization Mass Spectrometry, John Wiley and Sons, New York, 1997 Search PubMed.
- H. Z. Lian, W. H. Wang and D. N. Li, J. Sep. Sci., 2005, 28, 1179–1187 CrossRef CAS.
- X. Ming, S. Y. Han, Z. C. Qi, D. Sheng and H. Z. Lian, Talanta, 2009, 79, 752–761 CrossRef CAS PubMed.
- S. Y. Han, X. Ming, Z. C. Qi, D. Sheng and H. Z. Lian, Anal. Bioanal. Chem., 2010, 398, 2731–2743 CrossRef CAS PubMed.
- S. Y. Han, J. Q. Qiao, Y. Y. Zhang, H. Z. Lian and X. Ge, Talanta, 2012, 97, 355–361 CrossRef CAS PubMed.
- Z. C. Qi, S. Y. Han, Z. Y. Wu, F. Y. Chen, X. W. Cao, H. Z. Lian and L. Mao, Curr. Anal. Chem., 2014, 10, 172–181 CrossRef CAS.
- N. El Tayar, A. Tsantili-Kakoulidou, T. Roethlisberger, B. Testa and J. Gal, J. Chromatogr. A, 1988, 439, 237–244 CrossRef CAS.
- D. J. Minick, J. H. Frenz, M. A. Patrick and D. A. Brent, J. Med. Chem., 1988, 31, 1923–1933 CrossRef CAS.
- M. H. Abraham, H. J. Chadha and A. Leo, J. Chromatogr. A, 1994, 685, 203–219 CrossRef CAS.
- J. H. Park, J. J. Chae, T. H. Nah and M. D. Jang, J. Chromatogr. A, 1994, 664, 149–158 CrossRef CAS.
- R. Kaliszan, M. A. Van Straten, M. Markuszewski, C. A. Cramers and H. A. Claessens, J. Chromatogr. A, 1999, 855, 455–486 CrossRef CAS.
- F. Lombardo, M. Y. Shalaeva, K. A. Tupper, F. Gao and M. H. Abraham, J. Med. Chem., 2000, 43, 2922–2928 CrossRef CAS PubMed.
- X. Liu, H. Tanaka, A. Yamauchi, B. Testa and H. Chuman, Helv. Chim. Acta, 2004, 87, 2866–2876 CrossRef CAS.
- F. Lombardo, M. Y. Shalaeva, K. A. Tupper and F. Gao, J. Med. Chem., 2001, 44, 2490–2497 CrossRef CAS PubMed.
- C. Giaginis, S. Theocharis and A. Tsantili-Kakoulidou, J. Chromatogr. A, 2007, 1166, 116–125 CrossRef CAS PubMed.
- D. Dellis, C. Giaginis and A. Tsantili-Kakoulidou, J. Pharm. Biomed. Anal., 2007, 44, 57–62 CrossRef CAS PubMed.
- X. Liu, H. Tanaka, A. Yamauchi, B. Testa and H. Chuman, J. Chromatogr. A, 2005, 1091, 51–59 CrossRef CAS PubMed.
- C. Giaginis and A. Tsantili-Kakoulidou, J. Liq. Chromatogr. Relat. Technol., 2008, 31, 79–96 CrossRef CAS.
- T. Fujita, J. Iwasa and C. Hansch, J. Am. Chem. Soc., 1964, 86, 5175–5180 CrossRef CAS.
- H. Tomida, T. Yotsuyanagi and K. Ikeda, Himalayan Chem. Pharm. Bull., 1978, 26, 2824–2831 CrossRef CAS.
- K. Gerogi and K. S. Boos, Chromatographia, 2006, 63, 523–531 Search PubMed.
- O. E. Schultz, C. Jung and K. E. Z. Moller, Naturforscher, 1970, 25, 1024–1028 CAS.
- J. J. Sangster, J. Phys. Chem. Ref. Data, 1989, 18, 1111–1229 CrossRef CAS PubMed.
- C. Hansch and A. J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, Wiley, New York, 1979 Search PubMed.
- S. H. Unger, J. R. Cook and J. S. Hollenberg, J. Pharm. Sci., 1978, 67, 1364–1367 CrossRef CAS.
- T. Hanai, K. Koizumi, T. Kinoshita, R. Arora and F. Ahmed, J. Chromatogr. A, 1997, 762, 55–61 CrossRef CAS.
- K. Kishida and T. Otori, Jpn. J. Ophthalmol., 1980, 24, 251–259 CAS.
- J. G. Speight, Lange's Handbook of Chemistry, McGraw-Hill, New York, 16th edn, 2005 Search PubMed.
- A. I. Biggs and R. A. Robinson, J. Chem. Soc., 1961, 388–393 RSC.
- N. F. Hall and M. R. Sprinkle, J. Am. Chem. Soc., 1932, 54, 3469–3485 CrossRef CAS.
- K. Ong, B. Douglas and R. Robinson, J. Chem. Eng. Data, 1966, 11, 574–581 CrossRef CAS.
- M. M. Fickling, A. Fischer, B. R. Mann, J. Packer and J. Vaughan, J. Am. Chem. Soc., 1959, 81, 4226–4230 CrossRef CAS.
- H. Brown, E. Braude and F. Nachod, Determination of Organic Structures by Physical Methods, Academic Press, New York, 1955 Search PubMed.
- R. Andon, J. Cox and D. Herington, Trans. Faraday Soc., 1954, 30, 918–922 RSC.
- Y. Shabarov, V. Potapov and R. Levin, Russ. J. Gen. Chem., 1964, 34, 2865 Search PubMed.
- J. Flieger, J. Chromatogr. A, 2007, 1175, 207–216 CrossRef CAS PubMed.
- S. Y. Han, J. Q. Qiao, Y. Y. Zhang, L. L. Yang, H. Z. Lian, X. Ge and H. Y. Chen, Chemosphere, 2011, 83, 131–136 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |