
Curcumin induces structural change and reduces the growth of amyloid-β fibrils: a QCM-D study†
Abstract
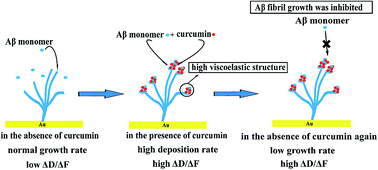
Amyloid-β (Aβ) fibrillation is a crucial factor in the etiology of Alzheimer's disease (AD). Curcumin has been widely studied and considered as a promising drug for AD treatment, but its molecular mechanism in inhibiting Aβ aggregation is still not clearly defined. In the present study, quartz crystal microbalance with dissipation monitoring (QCM-D) was used to analyse the growth behaviors of Aβ fibrils in the presence and absence of curcumin. The viscoelasticity properties that reflect the structural information of the resulting Aβ aggregates were also analysed using a ΔD/ΔF plot. It was found that the presence of curcumin could accelerate the deposition process of Aβ monomers on the initially immobilized fibrils, with the growth rate being 21.1–120.6% higher than that of the control. Viscoelasticity analysis showed that curcumin-induced Aβ aggregates exhibited a much more flexible structure than that of the initial fibrils, and the degree of this kind of structural conversion was related to the concentration of curcumin. Importantly, we found that further deposition of Aβ monomers on these loosely constructed Aβ aggregates was significantly inhibited, with the growth rate being only 34.2% of initial rate. Therefore, the QCM-D study demonstrated that curcumin inhibited the growth of Aβ fibrils through a process that led to the structural conversion of the growing fibrils, and directed these fibrils to an off-pathway aggregation, which may hinder the formation of long Aβ fibrils that could cause neuronal damage.
- This article is part of the themed collection: Towards understanding and treating Alzheimer’s disease
 Please wait while we load your content...
Please wait while we load your content...

