
Photo-polymerisable electrospun fibres of N-methacrylate glycol chitosan for biomedical applications†
Ilaria Romano*a,
Elisa Mele‡
*a,
José A. Heredia-Guerreroa,
Luca Ceseracciua,
Hadi Hajialiab,
Luca Goldonic,
Lara Marinia and
Athanassia Athanassiou*a
aSmart Materials, Nanophysics, Istituto Italiano di Tecnologia, Via Morego 30, 16163, Genoa, Italy. E-mail: ilaria.romano@iit.it; elisa.mele@iit.it; athanassia.athanassiou@iit.it
bDIBRIS, University of Genoa, Via Opera Pia 13, 16145, Genoa, Italy
cDrug Discovery and Development, Istituto Italiano di Tecnologia, Via Morego 30, 16163, Genoa, Italy
First published on 26th February 2015
Abstract
The availability of nanofibrous substrates with engineered properties, such as controlled porosity, mechanical conformability, biodegradation profile and drug release, is of strategic importance in the biomedical sector. Here, we demonstrate that N-methacrylate glycol chitosan, a photo-polymerisable, biocompatible and water-soluble derivative of chitosan, can be easily processed to create non-woven mats of nanofibres with controlled physicochemical characteristics. The produced fibrous mats are characterised by thermal stability, Young's modulus of 140 MPa and ultimate strength of 4 MPa. The degree of cross-linking of the realised fibres regulates their durability and degradation profile under conditions of high humidity, but also allows controlling the delivery over time of active agents encapsulated inside the fibres. We demonstrate that the N-methacrylate glycol chitosan nanofibres are able to release an antimicrobial drug within 24 hours. Moreover, cells proliferation of 85% indicates that non-cytotoxic substances were released from the electrospun mats.
Introduction
Scaffolding materials to be used in regenerative medicine and tissue engineering require characteristics of excellent biocompatibility, controlled biodegradability and engineered porosity.1–6 It is of paramount importance that the degradation time of these substrates is comparable to the time needed for the healing and/or the regeneration of the target tissue. Moreover, architectures with appropriate permeability are necessary to promote the diffusion of nutrients, the release of drugs, and the proliferation of cells.Photopolymerisable polymers derived from naturally-occurring polysaccharides represent an interesting class of biomaterials, whose biodegradation profile can be easily tuned by acting on their degree of crosslinking.7–9 To this purpose, chemically modified chitosan, a polymer obtained from the exoskeleton of arthropods or from the cell walls of fungi,10 has been extensively exploited. In particular, Amsden and co-workers synthesised a water soluble derivative of chitosan, N-methacrylate glycol chitosan (MGC), by modifying glycol chitosan with glycidyl methacrylate.11 MGC can be cross-linked by ultraviolet (UV) light, and its use has been demonstrated for cell encapsulation and drug delivery.12–16 One important property of the MGC hydrogel is the non-cytotoxicity that has been advantageously exploited in the realisation of injectable scaffolds for the regeneration of soft tissues.17–19
To the best of our knowledge, so far MGC has been only processed as bulk un-textured hydrogel, but not in the form of nanofibres. The availability of MGC nanofibrous substrates could significantly increase the potentialities of this photopolymerisable material in the biomedical sectors. In fact, non-woven mats of nanofibres, realised by the electrostatic spinning (ES) technique, possess high specific surface area, interconnected porosity and mechanical conformability. These features are essential in tissue regeneration for promoting the growth of cells since they mimic the extracellular matrix (EMC) architecture. Furthermore, they can guarantee efficient transport of nutrients and gases to the cells, but also controlled release of loaded bioactive compounds.20–22
Here, we show for the first time the fabrication of non-woven mats of long and well-defined nanofibres of N-methacrylate glycol chitosan by electrospinning. Acting on the cross-linking degree of the resulting MGC fibrous scaffolds using UV light, we regulated their degradability in moisture-rich atmospheres and effectively controlled the rate of release of encapsulated drugs. We simulated the moist environment of an open wound, and demonstrated that fibres mats of specific thickness were able to deliver antimicrobial agents in the time frame of 24 hours (typical time of replacement of a wound bandage), while their integrity was preserved in order to be easily managed and removed. The high flexibility offered by the here presented electrospun MGC supports makes them promising as biomedical devices.
Experimental
Chemicals and cell line
Glycol chitosan, dimethylsulfoxide (DMSO), the photoinitiator Irgacure 2959, glycidyl methacrylate, polyethylene oxide (PEO, Mw 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), sodium hydroxide, hydrochloric acid, agarose, deuterium oxide (DO), sodium deuteroxide (NaOD), Dulbecco's Modified Eagle's Medium (DMEM), Bovine Calf Serum (BCS), trypsin–EDTA solution, penicillin–streptomycin and thiazolyl blue tetrazolium bromide (MTT) were purchased from Sigma Aldrich. A water solution of eosin at 2%, known as Neomercurocromo®, was obtained from Laboratorio Farmaceutiche Specialità Igienico Terapeutiche S.I.T. s.r.l. Fibroblast cells from murine embryo (NIH3T3-cells) were obtained from ATCC® (CRL-1658™).
000 g mol−1), sodium hydroxide, hydrochloric acid, agarose, deuterium oxide (DO), sodium deuteroxide (NaOD), Dulbecco's Modified Eagle's Medium (DMEM), Bovine Calf Serum (BCS), trypsin–EDTA solution, penicillin–streptomycin and thiazolyl blue tetrazolium bromide (MTT) were purchased from Sigma Aldrich. A water solution of eosin at 2%, known as Neomercurocromo®, was obtained from Laboratorio Farmaceutiche Specialità Igienico Terapeutiche S.I.T. s.r.l. Fibroblast cells from murine embryo (NIH3T3-cells) were obtained from ATCC® (CRL-1658™).
Synthesis and characterisation of MGC
N-Methacrylate glycol chitosan was synthesised according to the procedure described in ref. 11. Briefly, the commercially available glycol chitosan was first purified by 50 kDa dialysis membrane against water for 48 hours. Then, an aqueous solution containing 1.9% w/v of the purified glycol chitosan was prepared, and adjusted with NaOH 1 M to reach pH 9.0. The solution was then reacted with glycidyl methacrylate for 24 hours to obtain a 3% degree of substitution (DOS) and neutralised with HCl 0.1 M. The resulting material was purified by 12 kDa dialysis membrane against water for 8 hours, and lyophilised for 2–3 days. In this way, a powder of N-methacrylate glycol chitosan with a degree of substitution (DOS) of 3% was obtained, as demonstrated by NMR analysis (Fig. S1†). 1H NMR spectra were acquired at 400.13 MHz with a spectrometer Bruker Avance 400 Ultra Shield TM Plus. The MGC powder was dissolved in DO at a concentration of 20 mg mL−1, and the resulting solution was adjusted with 0.1 M NaOD to reach a pH > 10. The samples were allowed to equilibrate for 10 minutes at 90 °C within the spectrometer prior to shimming. Before each acquisition the automatic matching and tuning was run and the homogeneity was automatically adjusted. 128 transients were accumulated by applying a 30 flip angle, 65K complex data points, with a spectral width of 20.5 ppm and a relaxation delay of 5 seconds. The temperature was actively controlled at 363 K. The free induction decay (FID) signals were multiplied by an exponential function equivalent to that of 0.5 Hz line-broadening factor and then Fourier transformed. Spectra were manually phased and automatically baseline corrected by means of an automatic alternate algorithm (Topspin 3.1, Bruker) and referred to the signal at 6.2 ppm, as reported by work presented by Amsden et al.15 Each integral was manually adjusted by a bias and a slope correction.Fabrication of the MGC fibrous mats
Water solutions consisting of 4% w/v of MGC and 1 or 5% w/v of photoinitiator Irgacure 2959 were prepared and used for electro-spinning. To facilitate the formation of the nanofibres, an optimised amount of polyethylene oxide (PEO) was added to the pristine MGC solution (MGC/PEO relative ratio of 7:3). As previously demonstrated by other groups, the majority of polysaccharides and their derivatives are hard to electrospin.23 For this reason, biocompatible polymers, like PEO, are typically used as additives.24,25 From now on any reference in the manuscript to MGC nanofibres would imply the blend MGC/PEO (7:3). In order to improve the uniformity of the nanofibres, we added DMSO as co-solvent to the initial MGC solutions (concentration of 10% w/v). DMSO plays a double role that is essential for the ES procedure of MGC. First, due to its high dielectric constant,26,27 it increases the electrical conductivity of the MGC solution, and second it loosens the chitosan chain entanglement.25
In our ES set-up, the fibres were extruded by applying a voltage of 10 kV (high voltage power supply EH40R2.5, Glassman High Voltage Inc.) between a stainless steel 23-gauge needle and an aluminium plate, placed at a relative distance of 20 cm. The solutions were injected through the needle by a syringe pump (NE-1000, New Era Pump Systems Inc.) working at a flow rate of 0.6 mL h−1. Immediately after production (without drying procedures), part of the collected fibres was cross-linked by using UV light at (405 ± 10) nm (20 mW cm−2, irradiation time of 30 minutes).
Characterisation of the nanofibres
The morphology and the size of the electrospun fibres were analysed by Scanning electron microscopy (SEM). A JEOL JSM-6490LA microscope working in high vacuum mode, with an acceleration voltage of 15 kV, was used. A coating of 10 nm Au/Pd was required to prevent charging effects. We investigated the chemical properties of the MGC fibres by an attenuated total reflectance (ATR) accessory (MIRacle ATR, PIKE Technologies) coupled to a Fourier transform infrared (FTIR) spectrometer (Equinox 70 FT-IR, Bruker). The spectra were recorded in the range from 3800 to 600 cm−1, with 4 cm−1 resolution (accumulating 128 scans).The mechanical properties of the scaffolds were characterised by uniaxial tension tests on a dual column universal testing machine (Instron 3365). Mats were cut as prismatic specimens with a width of 4 mm and an effective length of 25 mm. In order to avoid accidental damage during handling, the specimens were first taped on custom made paper frames composed by two detachable halves, then mounted on the measuring machine's hydraulic clamps. Displacement was applied with the rate of 1 mm min−1. From the resulting stress strain curves, the Young's modulus, the ultimate strength and the elongation at fracture were extracted. Measurements were performed on 5 samples.
The thermal degradation behaviour of the electrospun MGC/PEO nanofibres and of the pure components (MGC and PEO) was investigated by thermogravimetric (TGA) analysis using the Q500 analyser from TA Instruments. The measurements were performed under an inert N2 atmosphere with a flow rate of 50 mL min−1 in a temperature range of 30–600 °C at heating rate of 10 °C min−1.
The biocompatibility of the fibrous MGC scaffolds was evaluated by indirect cytotoxicity assays with immortalized mouse fibroblasts (NIH/3T3) as model cell line. After UV cross-linking and sterilization, the fibrous mats were immersed in the culture medium (DMEM) supplemented with 10% BCS and 1% antibiotics (100 U mL−1 penicillin and 0.1 mg mL−1 streptomycin) for 24 hours, in order to obtain their complete dissolution. In particular, according with the ISO10993-5 standard test method, a fibrous sample with an area of 6 cm2 was dissolved in 1 mL of DMEM. Meanwhile, NIH3T3 cells were seeded on a 96-well tissue culture polystyrene plate (TCPS) with a density of approximately 10000 cells per well, and cultured for 24 hours at 37 °C with 5% CO2 to allow the attachment. Then, the culture medium was replaced with the extraction one and the cells were incubated for other 24 hours.
Investigation of the fibre stability and drug release in aqueous environments
The fibrous samples (5% w/v of photoinitiator, thickness ∼ 300 μm) were stored in a box with controlled humidified atmosphere at 100% relative humidity (temperature of 25 °C), and their structural stability was analysed by SEM after fixed time intervals.As model compound, the antimicrobial Neomercurocromo® was incorporated inside the MGC fibres. A MGC:PEO solution containing 1 v/v% of Neomercurocromo was prepared and electrospun according to the procedure previously described. Composite nanofibrous mats (before and after UV irradiation) with 1 cm2 area (thickness ∼ 300 μm) were placed in contact with the surface of a thick film of agarose hydrogel (1% in water, pH 7). The diffusion of eosin through the hydrogel was recorded as a function of time by using a UV-visible spectrophotometer, Varian Cary 6000i, in absorbance mode from 200 to 800 nm. In particular, the agarose was previously gelified inside a cuvette, and the samples were gently deposited onto the hydrogel, before positioning the cuvette in the spectrophotometer. Then, the absorption spectra were collected regularly over time, from a portion of the gel located just underneath the fibrous mats. The experiments were performed in quintuplicate at room temperature.
Results and discussion
The morphological analysis, carried out by SEM, revealed that the electrospun mats of MGC with 1% photoinitiator mainly consisted of a dense network of nanofibres (Fig. 1a). However, the fibres' diameter appeared non-uniform throughout their length, since bead-like features were present. As clearly visible in Fig. 1b, the structure of the electrospun fibres was substantially improved by the addition of DMSO, since the presence of beads was strongly reduced. The resulting nanofibres showed cylindrical shape and regular morphology with an average diameter of (172 ± 31) nm. The nanofibres' size distribution was also quite homogeneous, since 80% of the population had diameters in the range of 150–210 nm (Fig. 1d). Well-defined nanofibres were also produced by increasing the amount of photoinitiator from 1 to 5% (Fig. 1c). In this case, the average diameter increased, becoming (266 ± 39) nm (Fig. 1d), and 40% of the population was monodispersed with a size of 230 nm. On the contrary, when the degree of acrylation of MGC was increased from 3 to 7%, we observed the formation of beaded fibrillar structures (Fig. S2†).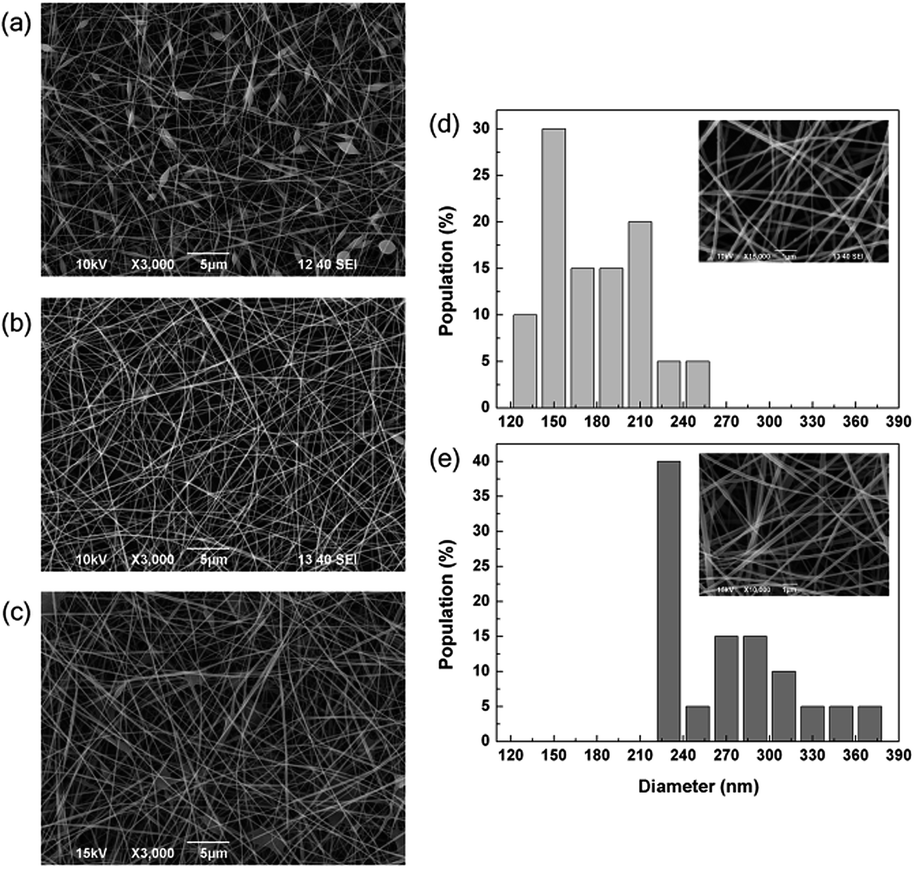 | ||
| Fig. 1 SEM images of the cross-linked fibrous networks, electrospun from MGC solutions containing (a) 1% w/v of photoinitiator, (b) 1% w/v of photoinitiator and 10% w/v of DMSO, (c) 5% w/v of photoinitiator and 10% w/v of DMSO. Corresponding size distribution of the realized nanofibres with (d) 1%, and (e) 5% of photoinitiator, and related high magnification SEM pictures in the insets. | ||
After production, the fibres were cross-linked using UV light. No appreciable differences were observed in the size and morphology of the nanofibres before and after light exposure. Indeed, the SEM pictures presented in Fig. 1 represent the cross-linked electrospun mats but they are identical to the just-electrospun ones (Fig. S3†). It is worth noting that, in the case of ES, the cross-linking process is not necessary for imparting a physical shape to the fibres. In fact, during ES complete evaporation of the solvent occurs and the nanofibres reach the collector in a dry, solid state, obtaining their final structure even before the UV exposure. The photo-cross-linking procedure solely affects their durability when they are exposed to humid environments, as will be discussed later. On the contrary, when MGC solutions are used for the production of bulk hydrogel scaffolds, the light irradiation is essential to structure the polymer in the desired form in order to be usable.19
As it is possible to observe from the SEM images, the fibres were homogeneously organised within the mats. This was also confirmed by mechanical testing that showed highly reproducible stress–strain curves (Fig. S4†). Values of Young's modulus and ultimate strength of (139 ± 6) MPa and (3.7 ± 0.6) MPa were measured, respectively, with an elongation at fracture of around 5%. Interestingly, the fracture proceeded without progressive inter-fibre shearing and tearing, indicating robustness and a well-connected structure of the mat. The reported values are comparable with those of chitosan electrospun mats that were cross-linked with other agents, such as glutaraldehyde (GA) vapour or epichlorohydrin.28 For instance, Schiffman and coworkers reported values of approximately 150 and 1 MPa for the Young's modulus and the tensile strength of GA-crosslinked chitosan mats, respectively.29 Therefore, MGC can be considered alternative to chitosan in the production of mechanically robust electrospun mats, particularly for biomedical applications that required the use of water soluble compounds.
The chemical properties of the electrospun samples were analysed by ATR-FTIR. As shown in Fig. 2a, in both just-electrospun and cross-linked nanofibres, the following bands associated to PEO are easily distinguishable: C–H stretching at 2880 cm−1, CH2 scissoring at 1466 cm−1, CH2 wagging at 1360 and 1340 cm−1, CH2 twisting at 1279 cm−1, C–O–C stretching at 1095 cm−1, CH2 rocking at 960 and 841 cm−1.25 Moreover, the following bands ascribed to glycol chitosan can be assigned: the overlap of O–H and N–H stretching vibrations at 3365 cm−1, the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–NHR stretching at 1670 cm−1 and the bending of non-acylated 2-amineglucose primary amines groups at 1595 cm−1.30,31 However, the vibrations of N-methacrylate residues were hardly observed, probably as consequence of their low amount in the final composition of the fibres. As demonstrated by Amsden and co-workers by nuclear magnetic resonance spectroscopy, the N-methacrylate residues are grafted on the glycol chitosan backbone via amine groups.11 In order to better characterise the chemical differences in the fibres before and after UV irradiation, we subtracted the spectrum of the cross-linked nanofibres from the one of the just-electrospun nanofibres (Fig. 2b and c). Positive absorptions were observed around 3365 (νO–H or νN–H), 1650 and 1595 cm−1 (δN–H). The vibration at 1650 cm−1 could be related to the stretching of CC from the methacrylate group.32 These positive differences in FT-IR signals are indicative of a loss of –OH, CC and amine functional groups after light irradiation. The opening of the double bonds and the reaction with electron-rich groups (hydroxyl and amine functional groups) can be related to the cross-linking of the chitosan polymer chains, as shown in the schematic representation in Fig. 2d.
O–NHR stretching at 1670 cm−1 and the bending of non-acylated 2-amineglucose primary amines groups at 1595 cm−1.30,31 However, the vibrations of N-methacrylate residues were hardly observed, probably as consequence of their low amount in the final composition of the fibres. As demonstrated by Amsden and co-workers by nuclear magnetic resonance spectroscopy, the N-methacrylate residues are grafted on the glycol chitosan backbone via amine groups.11 In order to better characterise the chemical differences in the fibres before and after UV irradiation, we subtracted the spectrum of the cross-linked nanofibres from the one of the just-electrospun nanofibres (Fig. 2b and c). Positive absorptions were observed around 3365 (νO–H or νN–H), 1650 and 1595 cm−1 (δN–H). The vibration at 1650 cm−1 could be related to the stretching of CC from the methacrylate group.32 These positive differences in FT-IR signals are indicative of a loss of –OH, CC and amine functional groups after light irradiation. The opening of the double bonds and the reaction with electron-rich groups (hydroxyl and amine functional groups) can be related to the cross-linking of the chitosan polymer chains, as shown in the schematic representation in Fig. 2d.
 | ||
| Fig. 2 (a) ATR-FTIR spectra of PEO and MGC nanofibres containing 5% of photoinitiator, before and after UV irradiation. The chemical structure of the N-methacrylate glycol chitosan is included in the inset. (b) and (c) Difference between the spectrum of the just-electrospun and cross-linked fibres in the spectral region of the 3650–3000 cm−1 and 1700–1550 cm−1, respectively. (d) Schematic representation of the polymer structure of the nanofibres after the UV irradiation. | ||
The structural stability of the MGC electrospun networks, before and after UV exposure, was investigated under specific conditions of humidity. After the observation of the samples by SEM, it was visible that after 1 hour in the moisture-rich environment the not-crosslinked fibres started fusing together (Fig. 3a), with their diameters getting bigger and the presence of large unstructured regions being visible. This was even more evident after 1 day of storage at the high humidity when a complete closure of the pores, previously existing in the electrospun mat, was observed. Indeed, from Fig. 3c it is clear that the fibrous network has been totally disappeared, substituted by a uniform MGC film. From the SEM analysis, we estimated that initially, in normal environmental conditions, the 72% of the sample surface area was occupied by the MGC fibres. Instead, after 1 hour in the humid environment, 90% of the surface area of the mat was covered by the polymer and the totality after 1 day. On the contrary, the cross-linked fibres were much more stable. In fact, they maintained their shape after 1 hour in the humid atmosphere almost entirely, with only few areas being flattened due to the moisture (Fig. 3b). In fact, in this case image processing showed that the nanofibres were present on the 77% of the surface area, likewise the as-prepared sample. It is most likely that the high water solubility of PEO played the main role in the observed topographical changes. After 1 day, the dimensions and the number of the flat regions appeared increased (93% of the surface area), with a consequent reduction of the porosity of the fibrous network. Nonetheless, the micrometer size pores, typical of the electrospun net, were still visible (Fig. 3d). By comparing the morphology of the just-electrospun and cross-linked MGC nanofibres after their storage at 100% RH, we can safely state that the cross-linking process allowed us to preserve the integrity of the mat after 24 hours (inset in Fig. 3d). This property is essential for wound managements that require the removal of the dressing after 1 day.
 | ||
| Fig. 3 SEM images showing the morphology of the electrospun MGC networks exposed to highly humid conditions for different time intervals: after 1 hour at 100% RH, (a) just-electrospun and (b) cross-linked fibres; after 1 day at 100% RH, (c) just-electrospun and (d) cross-linked fibres. Inset: photograph of the electrospun mat after 1 day at 100% RH. Scale bar: 0.5 cm. | ||
Together with structural stability under humid conditions, the produced MGC-PEO fibrous mats are also thermally stable in a range of temperature between 30–170 °C (TGA analysis in Fig. S5†). In fact, they started losing weight at around 200 °C, with two evident degradation steps at around 280 and 400 °C that are characteristic of pure MGC and PEO, respectively. Since materials for wound dressing typically operate at temperature lower than 40 °C, we can conclude that the produced MGC/PEO fibres can be applied in this sector without undergoing decomposition.
Moreover, indirect cytotoxicity assays revealed that the produced fibrous mats were biocompatible. To this end, immortalized mouse fibroblasts (NIH3T3) were used as model cell line, following the procedure described above. After cell incubation for 24 hours in the extraction medium (MGC fibres dissolved in DMEM), the adhered and proliferated cells were analysed by a colorimetric MTT assay. As shown in Fig. S6,† the cell proliferation was of about 85%, indication that the electrospun MGC-PEO fibres were non-cytotoxic and they did not release harmful substances to the living cells. Therefore, they can be potentially used for biomedical applications.
We observed that by acting on the cross-linking degree of the nanofibres we could control the release rate of active molecules encapsulated inside them. As model compound, a water solution containing 2% of eosin (Neomercurocromo®) and characterised by antimicrobial activity was incorporated inside the MGC nanofibres. The composite MGC–eosin nanofibrous mats were used for further tests with or without cross-linking. Agarose hydrogel plates were selected as systems to simulate the wet site of an open wound, due to their high water content.33 The diffusion of eosin through the hydrogel was directly related to its release from the fibres, and investigated by spectrophotometrical analysis. In Fig. 4, the intensity of the characteristic peak of eosin (at 540 nm) is plotted for the just-electrospun (black dots) and cross-linked fibres (white dots) at different time intervals. It can be observed that after 1 hour the intensity of the eosin peak increased to about 50% of its final value for the not cross-linked fibres, whereas it arrived to just 10% of its final value for the cross-linked ones. Therefore, the amount of eosin released from the not-irradiated samples in short times is much higher (burst release) than that released from the irradiated ones. For the not cross-linked fibrous substrates the measured absorption intensity arrived to its maximum value after 8 hours, indicating total release of the drug from the fibres. On the other hand, in the case of the photopolymerized MGC supports the maximum intensity of the eosin peak was achieved after 1 day. Similar release times have been reported for chitosan electrospun mats containing polyvinyl alcohol34 or poly(D,L-lactide-co-glycolide).35 In our case, we can also relate the release behaviour of the MGC mats to their degree of solubility when exposed to a moisture-rich environment, as presented above. Indeed, the 24 hours needed for the release of the eosin-compound from the cross-linked nanofibrous mat coincide with the time necessary to the same type of samples to alter their morphology in 100% RH environment but not to totally disintegrate. In order to dispose of dressings easier to handle we can further increase the thickness of the UV-treated electrospun fibres, without changing their drug release profile (inset in Fig. 4). This is very useful for daily wound management, when the dressing should be removed and changed after it has released the contained drug in a controlled manner throughout the 24 hours.
 | ||
| Fig. 4 Plot of the maximum intensity values for the absorption peak of eosin released from not- (black dots) and cross-linked (white dots) MGC fibres as function of time. Inset: photograph of MGC fibrous samples, after UV irradiation, containing 1% v/v of Neomercurocromo. Scale bar: 0.5 cm. | ||
Conclusions
The effective production of mechanically robust electrospun nanofibres of a photo-crosslinkable biomaterial, N-methacrylate glycol chitosan, was demonstrated. We overcame the difficulties frequently encountered when the ES technique is applied to reticulated polymers and natural polysaccharides, including other chitosan derivatives. Thanks to the process optimization, we were able to control over time the degradation profile of the fibres and the release rate of the encapsulated hydrophilic drugs. MGC nanofibres can have potential applications in diverse biomedical sectors, including tissue regeneration and wound dressing systems.Acknowledgements
José A. Heredia-Guerrero is supported by a Marie Curie Intra-European Fellowship (BIOPROTO project), financed by the EU's Seventh Framework Programme for Research (FP7).References
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS PubMed.
- G. E. Luckachan and C. K. S. Pillai, J. Polym. Environ., 2011, 19, 637–676 CrossRef CAS PubMed.
- G. G. Genchi, G. Ciofani, A. Polini, I. Liakos, D. Iandolo, A. Athanassiou, D. Pisignano, V. Mattoli and A. Menciassi, J. Tissue Eng. Regener. Med., 2012, 1932–6254 Search PubMed.
- S. Beke, F. Anjum, H. Tsushima, L. Ceseracciu, E. Chieregatti, A. Diaspro, A. Athanassiou and F. Brandi, J. R. Soc., Interface, 2012, 9, 3017–3026 CrossRef CAS PubMed.
- F. Brandi, F. Anjum, L. Ceseracciu, A. C. Barone and A. Athanassiou, J. Micromech. Microeng., 2011, 21, 054007 CrossRef.
- J. F. Mano, G. A. Silva, H. S. Azevedo, P. B. Malafaya, R. A. Sousa, S. S. Silva, L. F. Boesel, J. M. Oliveira, T. C. Santos, A. P. Marques, N. M. Neves and R. L. Reis, J. R. Soc., Interface, 2007, 4, 999–1030 CrossRef CAS PubMed.
- J. L. Ifkovits and J. A. Burdick, Tissue Eng., 2007, 13, 2369–2385 CrossRef CAS PubMed.
- O. Jeon, K. H. Bouhadir, J. M. Mansour and E. Alsberg, Biomaterials, 2009, 30, 2724–2734 CrossRef CAS PubMed.
- J. A. Burdick, C. Chung, X. Jia, M. A. Randolph and R. Langer, Biomacromolecules, 2005, 6, 386–391 CrossRef CAS PubMed.
- R. Jayakumara, D. Menona, K. Manzoora, S. V. Naira and H. Tamura, Carbohydr. Polym., 2010, 82, 227–232 CrossRef PubMed.
- B. G. Amsden, A. Sukarto, D. K. Knight and S. N. Shapka, Biomacromolecules, 2007, 8, 3758–3766 CrossRef CAS PubMed.
- J.-P. Chen, G.-Y. Chang and J.-K. Chen, Colloids Surf., A, 2008, 313–314, 183–188 CrossRef PubMed.
- M. Abrigo, S. L. McArthur and P. Kingshott, Macromol. Biosci., 2014, 14, 772–792 CrossRef CAS PubMed.
- L. Martinová and D. Lubasová, Res. J. Text. Apparel, 2008, 12, 72–79 Search PubMed.
- Z.-M. Huang, Y.-Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- J. W. S. Hayami, S. D. Waldman and B. G. Amsden, Macromol. Biosci., 2011, 11, 1672–1683 CrossRef CAS PubMed.
- A. Sukarto, C. Yu, L. E. Flynn and B. G. Amsden, Biomacromolecules, 2012, 13, 2490–2502 CrossRef CAS PubMed.
- J. W. S. Hayami, S. D. Waldman and B. G. Amsden, Biomacromolecules, 2013, 14, 4236–4247 CrossRef CAS PubMed.
- H. K. Cheung, T. T. Y. Han, D. M. Marecak, J. F. Watkins, B. G. Amsden and L. E. Flynn, Biomaterials, 2014, 35, 1914–1923 CrossRef CAS PubMed.
- M. Ignatova, N. Manolova and I. Rashkov, Macromol. Biosci., 2013, 13, 860–872 CrossRef CAS PubMed.
- P. D. Dalton, C. Vaquette, B. L. Farrugia, T. R. Dargaville, T. D. Brown and D. W. Hutmacher, Biomater. Sci., 2013, 1, 171–185 RSC.
- W. Liu, S. Thomopoulos and Y. Xia, Adv. Healthcare Mater., 2012, 1, 10–25 CrossRef CAS PubMed.
- A. C. Stijnman, I. Bodnar and R. H. Tromp, Food Hydrocolloids, 2011, 25, 1393–1398 CrossRef CAS PubMed.
- C. A. Bonino, K. Efimenko, S. I. Jeong, M. D. Krebs, E. Alsberg and S. A. Khan, Small, 2012, 8, 1928–1936 CrossRef CAS PubMed.
- N. Bhattarai, D. Edmondson, O. Veiseh, F. A. Matsen and M. Zhang, Biomaterials, 2005, 26, 6176–6184 CrossRef CAS PubMed.
- N. Choktaweesap, K. Arayanarakul, D. Aht-Ong, C. Meechaisue and P. Supaphol, Polym. J., 2007, 39, 622–631 CrossRef CAS.
- H. Jiang, D. Fang, B. S. Hsiao, B. Chu and W. Chen, Biomacromolecules, 2004, 5, 326–333 CrossRef CAS PubMed.
- A. E. Donius, M. A. Kiechel, C. L. Schauer and U. G. K. Wegst, J. R. Soc., Interface, 2013, 10, 20120946 CrossRef PubMed.
- J. D. Schiffman and C. L. Schauer, Biomacromolecules, 2007, 8, 594–601 CrossRef CAS PubMed.
- Z. Osman and A. K. Arof, Electrochim. Acta, 2003, 48, 993–999 CrossRef CAS.
- J. Xu, S. P. McCarthy, R. A. Gross and D. L. Kaplan, Macromolecules, 1996, 29, 3436–3440 CrossRef CAS.
- L. J. Bellamy, The Infrared Spectra of Complex Molecules, Chapman and Hall, New York, 1975 Search PubMed.
- B. G. Johnansson, Scand. J. Clin. Lab. Invest., 1972, 29, 7–19 CrossRef.
- W. Samprasit, R. Kaomongkolgit, M. Sukma, T. Rojanarata, T. Ngawhirunpat and P. Opanasopit, Carbohydr. Polym., 2015, 117, 933–940 CrossRef CAS PubMed.
- Z. X. Meng, W. Zheng, L. Li and Y. F. Zheng, Mater. Chem. Phys., 2011, 125, 606–611 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra02301g |
| ‡ Present address: Department of Materials, Loughborough University, Loughborough, Leicestershire, LE11 3TU, UK, E-mail: E-mail: E.Mele3@lboro.ac.uk |
| This journal is © The Royal Society of Chemistry 2015 |