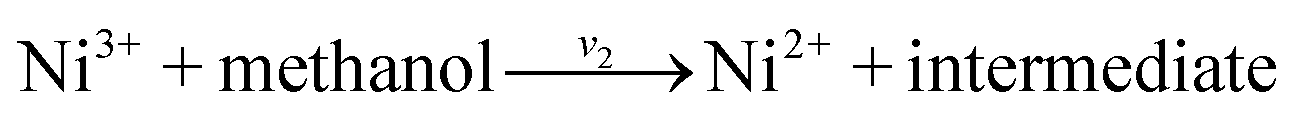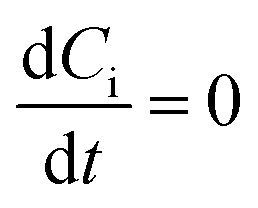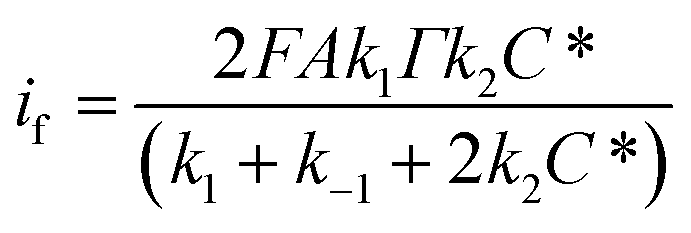DOI:
10.1039/C5RA02297E
(Paper)
RSC Adv., 2015,
5, 30394-30404
Physioelectrochemical and DFT investigation of metal oxide/p-type conductive polymer nanoparticles as an efficient catalyst for the electrocatalytic oxidation of methanol
Received
9th February 2015
, Accepted 18th March 2015
First published on 20th March 2015
Abstract
In the present study, in order to achieve an inexpensive tolerable anode catalyst for direct electrooxidation of methanol, poly tyramine nanoparticles and NiO, identified by PTy–NiO, were prepared by in situ electropolymerization of tyramine in the presence of an anionic surfactant under ultrasonic irradiation. Ni(II) ions are incorporated into the electrode by immersion of the polymeric modified electrode containing amine groups in a Ni(II) ion solution and a PTy–NiO composite film is formed in the alkaline medium on the surface of the electrode. The surface morphology of the film is studied by surface microscopy techniques. The presence of nickel in the films is confirmed by energy dispersive spectroscopy (EDS) analysis. Molecular modeling calculations are carried out for polymer–Ni with density functional theory (DFT) using a 6-31G(d,p) basis set and Gaussian 03 program package. Furthermore, the experimental absorbance spectra of PTy–NiO are compared with the results obtained from Maxwell Garnett theory (MGT) and the value of energy Eg = 3.75 eV of the composite obtained. Modified graphite electrodes (G/PTy–NiO and G/PTy–SDS–NiO) are examined for their redox processes and electrocatalytic activities towards the oxidation of methanol in alkaline solutions. Compared with G/PTy–NiO, the G/PTy–SDS–NiO electrode shows a higher catalytic performance in the electrocatalytic oxidation of methanol. A kinetic model was developed and the kinetic parameters were calculated using the methanol concentration dependency of charge transfer resistance derived from the impedance studies.
1. Introduction
In recent years, electrooxidation of small organic molecules has attracted much attention due to the development of direct liquid fuel cells, which require highly reactive fuels with high energy density. Methanol is one of the interesting future fuels for fuel cell applications. Compared with other cells, the direct methanol fuel cell (DMFC) has several advantages such as high efficiency, very low polluting emissions, a potentially renewable fuel source, fast and convenient refueling, simple operation and ease of fuel storage and distribution. The low operation temperature of a DMFC (typically <95 °C) allows for easy start up and rapid response to changes in load or operating conditions.1,2 However, compared to hydrogen based fuel cells, DMFC still have to be further developed. One of the problems still unsolved is the slow kinetics of methanol oxidation at the fuel cell’s anode.3 Considerable effort has been directed towards the study of methanol electrooxidation at high pH. The use of alkaline solutions in a fuel cell has many advantages such as increasing its efficiency,4,5 a wider selection of possible electrode materials, a higher efficiency of both anodic and cathodic processes, almost no sensitivity to the surface structure6 and negligible poisoning effects in alkaline solutions were observed.7,8
In the electrochemical oxidation of methanol, the electrode material is clearly an important factor where a highly efficient electrocatalyst is needed. As described previously,9,10 a considerable increase in power density and fuel utilization was obtained by optimizing different components of fuel cells. Different electrode materials based on Pt and Pt-binary electrodes were commonly used as catalysts for the electrochemical oxidation of methanol. As catalysis occurs at the surface, the catalyst needs to have the highest possible surface area. Carbon is a common choice for supporting nanosized electrocatalyst particles in DMFCs because of its large surface area, high electrical conductivity, and pore structure.11,12 However, this inert material (carbon) does not help electrocatalytic activity, but serves only as a mechanical support. Therefore, carbon-supported electrodes are generally used as catalysts,11 such as Pt–Ru and Pt–Ru–P/carbon nanocomposites,12 Pt/Ni and Pt/Ru/Ni alloy nanoparticles.13 It is well established that nickel can be used as a catalyst due to its surface oxidation properties. Many electrodes involving nickel have been used as catalysts in fuel cells. Ni has commonly been used as an electrocatalyst for both anodic and cathodic reactions in organic synthesis and water electrolysis. One of the very important uses of nickel as a catalyst is for the oxidation of alcohols. Several studies on the electrooxidation of alcohols on Ni have been reported.14–23 The study of conducting polymer modified electrodes is motivated primarily from the anticipation of a synergistic electrocatalytic benefit from the very good conducting and mechanical properties and their good adhesion to the electrode substrate. Due to their conductivity and stability, both in air and aqueous solution, polypyrrole, polyaniline, polyacetylene, polythiophene, polyindole, poly ortho-aminophenol and polythionine have been extensively studied for application in supercapacitors, sensors and catalysis.24–31 Moreover, the possibility of dispersing metallic particles inside the polymers gives electrodes with higher surface areas, enhanced electrocatalytic activities and facilitates electron transfer due to semiconducting properties29 and a change in the Fermi level towards the oxidation of organic molecules.24,28 Organic–inorganic composites have attracted considerable attention as they can combine the advantages of both components and may offer special properties through reinforcing or modifying each other.28 Electrodeposition is an effective way to make composite films with a large variety of tunable parameters and so has the advantage of convenient film control.
The purpose of the present work is the preparation of new a inexpensive tolerable composite catalyst, characterisation by optical modeling and DFT calculation and to study the electrocatalytic oxidation of methanol aiming at elucidation of the mechanism, derivation of the kinetic parameters of the process and the usefulness of the electrocatalytic process.
2. Computational studies
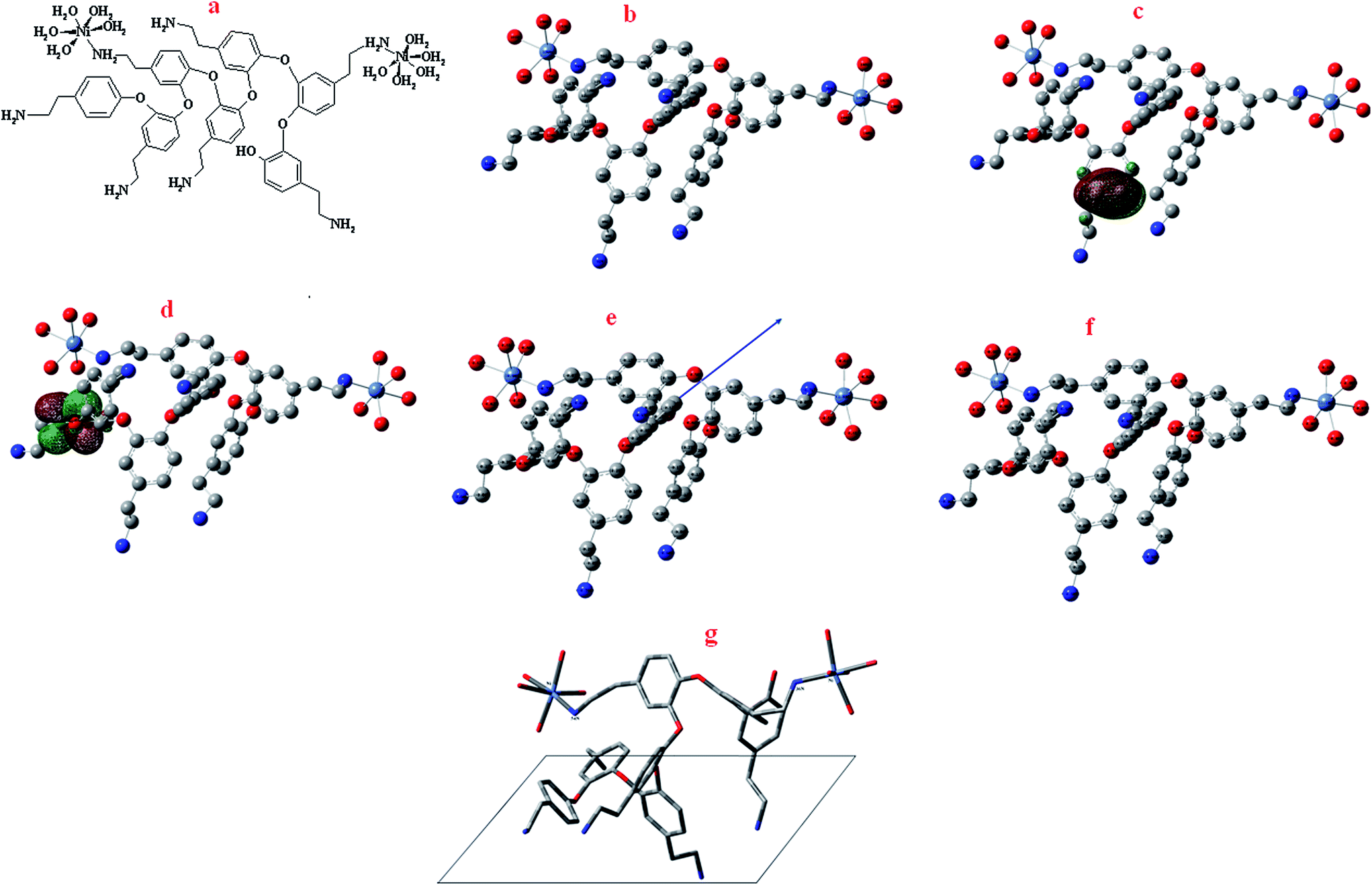
Computational study has been applied to present the optimized structure of the polymer/Ni complex. For phenol derivatives with amino groups, the reported voltammetric studies are interpreted analogy with to well-established aniline oxidation, i.e. a E(CE)n mechanism; by which the oxidation of o-aminophenol produces a ladder-structured film and reactive intermediates of 2-amino-phenoxazin-3-1 are formed in solution. In the case of tyramine (4-(2-aminoethyl)phenol) (Ty), because the amino function is separated from the phenolic ring by two methylene groups, it is expected that only the phenol moiety will be oxidized to perform the polymerization.32 In order to obtain valuable parameters concerning the polymerization of tyramine and complex formation with Ni, the polymerization of tyramine was revisited through quantum mechanical DFT calculations. According to EDS results concerning the percentage of Ni in the poly tyramine composite, [Ni2heptamer]4+ has been selected for optimization. Theoretical calculations were carried out at density functional theory (DFT) level using the 6-31G(d,p) basis set for all atoms with the Gaussian 03 program package for the Ni-heptamer. Electronic properties such as highest occupied molecular orbital (HOMO) energy, lowest unoccupied molecular orbital (LUMO) energy and frontier molecular orbital coefficients have been calculated. The molecular sketches of all compounds were drawn using Gauss View 03.33 Natural bond orbital (NBO) analysis was applied to determine the atomic charges of the Ni-heptamer. The results are presented in Fig. 1 and Table 1 and 2. The obtained results in Fig. 1 and Table 1 confirmed that the oxidizing phenol moiety performed the polymerization of tyramine. On the contrary, tyramine has a high electronic density in the phenol moiety related to the –NH2 group. Therefore, dimers can be formed through attacking the cation radical at that position and complex formation with Ni ions occurred from interaction between –NH2 groups and Ni ions. Such behavior provides a broad surface with electrostatic anchoring sites, where possible covalent bond formation with ethylamine groups are allowed. Ethylamine groups are very versatile in chemical and conformational senses. A polymer with such characteristics may be used for amplification of the electrochemical response in electrocatalytic processes. The schematic representation of the adsorption behavior of [Ni2heptamer]4+ on the surface of the electrode is presented in Fig. 1g.
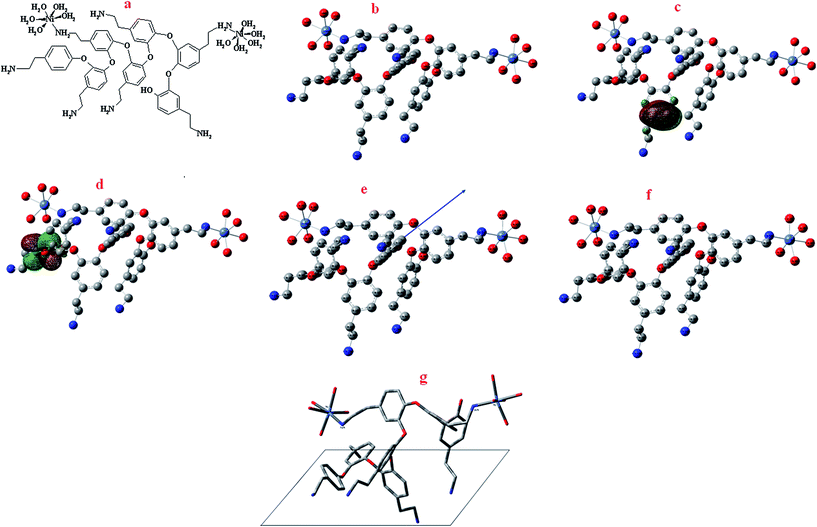 |
| | Fig. 1 (a) Structure of [Ni2heptamer]4+; (b) optimized molecular structure of [Ni2heptamer]4+, H atoms have been omitted for clarity; (c) the highest occupied molecular orbital (HOMO) of [Ni2heptamer]4+; (d) the lowest unoccupied molecular orbital (LUMO) of [Ni2heptamer]4+; (e) Mulliken charge population analysis and vector of the dipole moment of [Ni2heptamer]4+; (f) natural charge population analysis of [Ni2heptamer]4+ and (g) the schematic representation of the adsorption behavior of [Ni2heptamer]4+ on the surface of the electrode. | |
Table 1 Orbital energies for HOMO, LUMO, HOMO–LUMO gap energy (ΔE) and dipole moment (μ) of the compound in the gaseous phase. All quantum chemical parameters were calculated at DFT level using the 6-31G(d,p) basis set
| Compound |
EHOMO (eV) |
ELUMO (eV) |
ΔE (eV) |
μ (D) |
| [Ni2heptamer]4+ |
−10.840 |
−9.077 |
1.767 |
50.6378 |
Table 2 Mulliken and natural charges (e) for atoms
| Atom |
17N |
36N |
54N |
73N |
92N |
111N |
131N |
10O |
136Ni |
137Ni |
| Mulliken |
−0.606 |
−0.717 |
−0.732 |
−0.631 |
−0.594 |
−0.604 |
−0.610 |
−0.569 |
0.993 |
1.030 |
| Natural |
−0.921 |
−0.953 |
−0.951 |
−0.923 |
−0.905 |
−0.914 |
−0.925 |
−0.697 |
1.180 |
1.192 |
3. Optical modeling
In accordance with our previous work34–37 optical modeling has been applied for characterization of the composite film. We consider a region 0 ≤ z ≤ d to be occupied by NiO particles dispersed in the polymer (PTy) (Fig. 2) while the regions z ≤ 0 and z ≥ d are vacuous. Suppose an arbitrarily polarized plane wave is normally incident (axial excitation) on the chosen structure from z ≤ 0. The phasors of the incident, reflected and transmitted electric fields are given as:37–39| |
 | (1) |
where (as,ap), (rs,rp) and (ts,tp) are the amplitudes of the incident plane wave, and reflected and transmitted waves with S- or P-polarizations,  is the free space wave number, λ0 is the free space wavelength, ε0 = 8.854 × 10−12 F m−1 and μ0 = 4π × 10−7 H m−1 are the permittivity and permeability of free space (vacuum), respectively and
is the free space wave number, λ0 is the free space wavelength, ε0 = 8.854 × 10−12 F m−1 and μ0 = 4π × 10−7 H m−1 are the permittivity and permeability of free space (vacuum), respectively and ![[u with combining low line]](https://www.rsc.org/images/entities/i_char_0075_0332.gif) x,y,z are the unit vectors in Cartesian coordinates.
x,y,z are the unit vectors in Cartesian coordinates.
 |
| | Fig. 2 Schematic of the boundary-value problem for optical modeling of the NiO/PTy composite film. | |
The reflectance and transmittance amplitudes can be calculated using the continuity of the tangential components of electrical and magnetic fields at interfaces and solving the algebraic matrix equation:
| |
 | (2) |
The different terms and parameters of this equation are given in detail by Lakhtakia.38 Herein, the absorbance for natural light is determined as  , where
, where .
.
4. Experimental
Tyramine, sodium hydroxide, lithium perchlorate, nickel nitrate and methanol used in this work were Merck products of analytical grade and were used without further purification. All electrochemical experiments were carried out by an Autolab General Purpose System PGSTAT 30 (Eco-chime, Netherlands). A saturated calomel electrode (SCE), a Pt wire and a graphite (G) electrode (0.22 cm2) were used as the reference, counter and working electrodes, respectively. All studies were carried out at 298 ± 2 K. The microstructure of the electrodeposited composite was studied using different techniques. Scanning electron microscopy (SEM) was performed by a Tescan Vega II HiVac instrument. Transmission electron microscopy (TEM) was performed using a CEM 902A ZEISS transmission electron microscope, with an accelerating voltage of 80 kV.
Films of poly tyramine were formed on the graphite surface using a tyramine monomer solution (0.01 M tyramine in 0.1 M LiClO4 and 0.1 M HClO4) and (0.01 M tyramine in 0.1 mol L−1 LiClO4, 0.1 mol L−1 HClO4 and 0.005 mol L−1 sodium dodecyl sulfate (SDS)). The electropolymerization was carried out by potential cycling (40 cycles at a scan rate of 50 mV s−1 between −0.2 and 0.9 V versus SCE) under ultrasonic irradiation. In order to incorporate Ni(II) ions into the PTy film, the freshly electropolymerized graphite electrode was placed at open circuit in a well stirred aqueous solution of 0.1 mol L−1 Ni(NO3)2. Nickel was accumulated by complex formation between Ni(II) in solution and amine sites in the polymer backbone30,31 in a given period of time. The polarization behavior was examined in 1.0 mol L−1 NaOH for G/PTy–NiO using cyclic voltammetry. This technique allows oxide film formation in parallel with inspecting the electrochemical reactivity of the surface. Voltammograms were recorded by cycling the potential between 0.1 and 0.8 V at 100 mV s−1 until a stable voltammogram was obtained.
5. Results and discussion
In previous publications,24,25 we described how poly ortho-aminophenol films were electropolymerized in situ on the surface of a graphite electrode using cyclic voltammetry methods. Fig. 3a shows the typical multi-sweep cyclic voltammograms during Ty electropolymerization in the absence of SDS. As seen in Fig. 3a, tyramine (Ty) is oxidized irreversibly at around 650 mV without corresponding cathodic processes in the reverse scan. During the next cycles, one redox peak appears at lower potential, and its current does not considerably increase with potential cycling. This occurred because the soluble products produced on the surface of the electrode did not allow the monomer to reach the electrode and produce more monocation radicals. Therefore, a longer potential cycling time is needed for transformation to PTy.
 |
| | Fig. 3 The typical multi-sweep cyclic voltammograms during electropolymerization of Ty in the absence of SDS (a) and in the presence of SDS (b) and comparison (c). | |
By adding 0.005 mol L−1 of SDS anionic surfactant to the monomer solution, the monomer oxidation potential was shifted and its oxidation current increased (Fig. 3b). Moreover, the rate of polymerization increased considerably and the redox peaks due to PTy grew (Fig. 3c). Furthermore, under successive potential cycling, their peak currents increased and their growth continued. These results show that, in the presence of SDS, the monomer can easily reach the electrode surface and produce more monocation radicals. In the case of Ty electropolymerization, the radical cation of the Ty monomer is formed on the electrode surface by oxidation of the monomer. This process is considered to be the rate determining step. NiO was formed in the Ni/polymer backbone after consecutive cycling in the NaOH solution which is described in the experimental section.
Fig. 4 shows the SEM and TEM images and EDS spectrum of the electrosynthesized electroactive film on the surface of the working electrode under ultrasonic irradiation. Nanoparticles of the electroactive polymer in the range of 20–50 nm have been shown in the SEM and TEM micrographs. Formation of the polymer nanoparticles has been related to ultrasonic irradiation effects during electropolymerization of the monomer. The incorporation of Ni in the poly tyramine film is confirmed by EDS analysis, which shows the presence of Ni located at 7.50, and 8.30 keV (Fig. 4). Also, the EDS spectrum of the electrochemically prepared film shows a signal of carbon (C) at 0.25 keV and oxygen (O) at 0.53 keV, characteristic of the PTy polymer. Because gold is used as a coating in the mounted electrode in SEM analysis we also observed two peaks at 2.07 and 9.8 keV of gold. Thus, the content of Ni (10.41%) in the film is confirmed by EDS analysis. For showing the high active surface area of the conductive polymer modified electrode the active surface area of bare graphite and the conductive polymer modified graphite electrode were compared by an electrochemical probe. Fig. 5 presents the cyclic voltammograms of the bare and modified graphite electrodes in a solution of 0.2 M NaNO3 containing 0.002 M [Fe(CN)6]3− recorded at a potential sweep rate of 100 mV s−1. Here, the electrochemically reversible redox couple ferri/ferrocyanide was used as a suitable probe to examine the active surface area of the films. For analytical purpose, it was assumed that the diffusion of oxidized, [Fe(CN)6]3−, and reduced, [Fe(CN)6]4−, species in the electrolyte is semi-infinite and one dimensional. The solution initially contains only oxidized species, the bulk concentration of these species being constantly away from the electrode, while the corresponding concentration of reduced species is zero. The supporting electrolyte, NaNO3, is assumed to present a sufficient concentration so that the contribution of the redox couple to migration can be neglected. Since the PTy synthesized in acidic solution is electroactive when the pH of the medium is less than 4 and is electroinactive when the pH is neutral or basic, under the experimental conditions used, ipeak was background corrected so that the only reversible reaction occurring with the redox couple is
| | |
[Fe(CN)6]3− + e− ⇄ [Fe(CN)6]4−
| (3) |
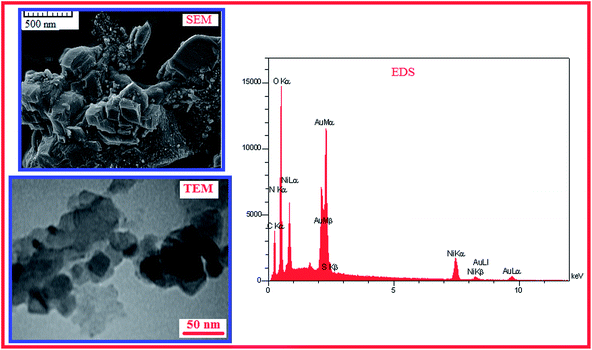 |
| | Fig. 4 SEM and TEM images and EDS spectrum of the G/PTy–SDS–NiO electrode. | |
 |
| | Fig. 5 Cyclic voltammograms for (1) bare graphite and (2) conductive polymer modified electrodes in a solution of 0.2 M NaNO3 containing 0.002 M [Fe(CN)6]3−, were recorded at a potential sweep rate of 100 mV s−1. | |
The high peak current of this redox couple at the modified electrode can be related to the high surface area of the electroactive polymer.
It is well known that in order to obtain an effective dielectric constant for dispersed phase volume fractions smaller than π/6 and a size of features much smaller than the wavelength of the incident electromagnetic wave in a two-component composite medium, we can use Maxwell Garnett theory (MGT).40 In this approximation method, the effective dielectric constant of a composite is defined as  for NiO particles embedded in PTy, where εPty and εNiO are the dielectric constants of PTy and NiO, while f is the volume fraction of dispersed particles. We have used the bulk experimental refractive indexes of NiO for homogenization.41 In our work, we extracted the refractive indexes k and n of PTy from the absorption coefficient and measured transmittance k = α
for NiO particles embedded in PTy, where εPty and εNiO are the dielectric constants of PTy and NiO, while f is the volume fraction of dispersed particles. We have used the bulk experimental refractive indexes of NiO for homogenization.41 In our work, we extracted the refractive indexes k and n of PTy from the absorption coefficient and measured transmittance k = α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) λ/4π, using the Swanepoel model,42,43 as shown in Fig. 6. The calculated absorbance spectrum as a function of wavelength for NiO particles embedded in PTy is depicted in Fig. 6c. In our calculation for Fig. 6, the structural parameters such as the thickness of the composite, the volume fraction of NiO particles, and fNiO = 0.1041, respectively, were fixed from the experimental results. In Fig. 6c, it can be observed that our optical modeling is consistent with the experimental results. The plot of (αhν)2 vs. photon energy is shown in Fig. 6d. The absorption coefficient was calculated from the absorbance spectra using the formula
λ/4π, using the Swanepoel model,42,43 as shown in Fig. 6. The calculated absorbance spectrum as a function of wavelength for NiO particles embedded in PTy is depicted in Fig. 6c. In our calculation for Fig. 6, the structural parameters such as the thickness of the composite, the volume fraction of NiO particles, and fNiO = 0.1041, respectively, were fixed from the experimental results. In Fig. 6c, it can be observed that our optical modeling is consistent with the experimental results. The plot of (αhν)2 vs. photon energy is shown in Fig. 6d. The absorption coefficient was calculated from the absorbance spectra using the formula  , where A is absorbance and hν is photon energy. It has been observed that the plot of (αhν)2 versus hν is linear over a wide range of photon energies indicating a direct type of transition. The intercept (extrapolation) of this plot (straight line) on the energy axis gives the energy band gap. The value of energy, Eg, of the composite was determined as Eg = 3.625 eV.
, where A is absorbance and hν is photon energy. It has been observed that the plot of (αhν)2 versus hν is linear over a wide range of photon energies indicating a direct type of transition. The intercept (extrapolation) of this plot (straight line) on the energy axis gives the energy band gap. The value of energy, Eg, of the composite was determined as Eg = 3.625 eV.
 |
| | Fig. 6 (a) Measured transmittance of PTy; (b) extracted refractive index and extinction coefficient for PTy as a function of wavelength; (c) calculated absorbance and (αhν)2 for NiO particles dispersed in PTy, absorbance vs. wavelength; (d) (αhν)2 vs. photon energy. | |
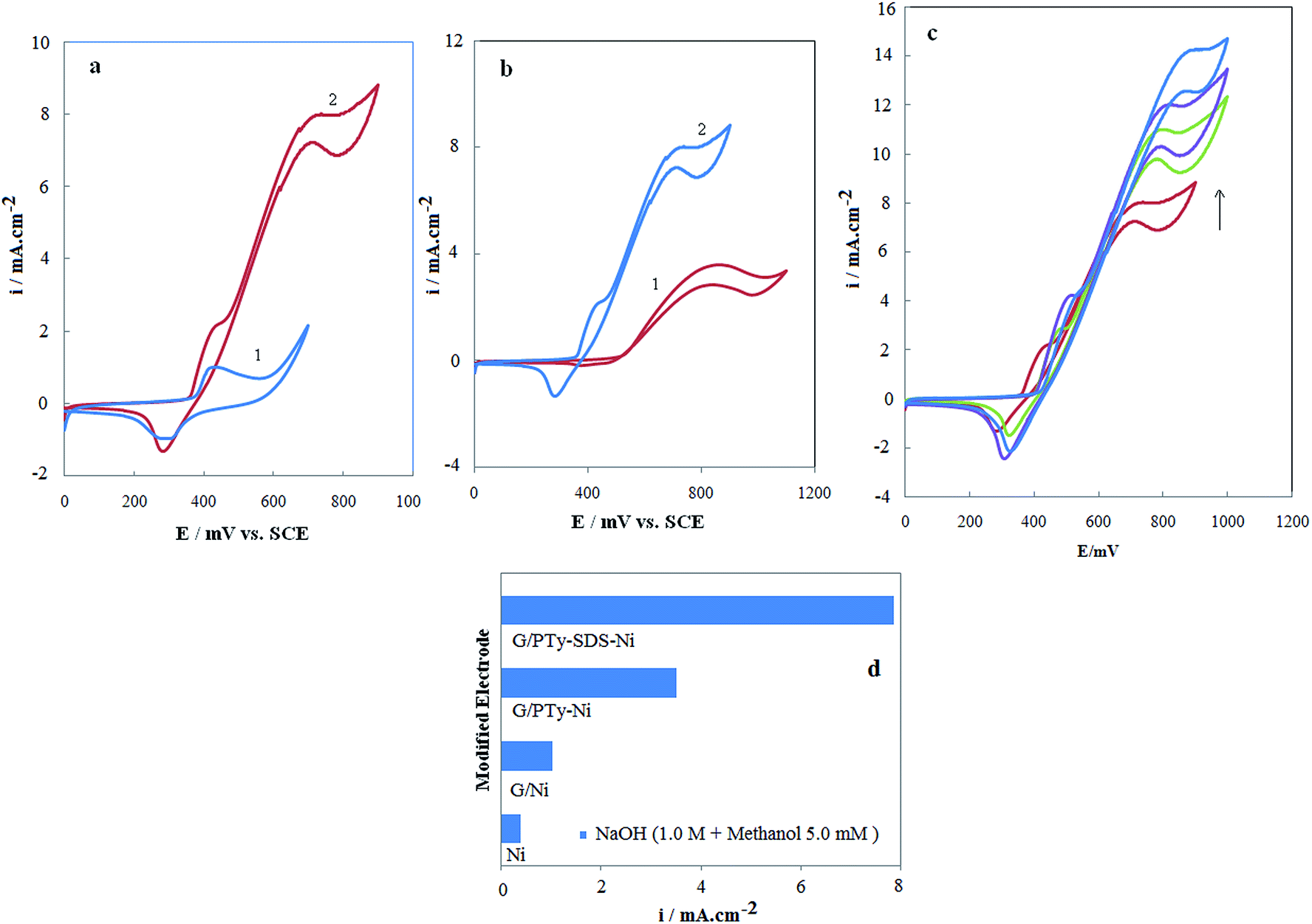
Fig. 7a shows cyclic voltammograms of the G/PTy–SDS–NiO electrode in 1 mol L−1 NaOH solution in the absence and presence of 0.005 mol L−1 methanol at a potential sweep rate of 10 mV s−1. As can be seen in 0.005 mol L−1 methanol the G/PTy–SDS–NiO electrode generates a higher current density for electrooxidation in NaOH solution. The larger methanol response at the G/PTy–SDS–NiO electrode is due to the composite material which enhances the catalytic properties of nickel oxide through fine dispersion of the catalyst particles in the conductive polymer matrix which results in a drastic increase in surface area. The electrocatalytic behavior of any material depends on various factors such as: (i) the position of the energy levels of the reactive species and the electrode material; (ii) the charge-transfer process across the electrode/electrolyte interface; (iii) the diffusion of the reactants into/near the electrode surface; and the surface morphology of the electrode. These results may be explained on the basis of electrochemical reactions at a semiconductor electrode. Here, PTy is considered as a p-type semiconductor. The impurity doping level will be situated above the uppermost valence level since PTy is a p-type material. Since the electrode is in an anodic condition, electrons are abstracted from the electrolyte and these combine with holes in the semiconductor. In the present study, it is therefore clear that charge transfer at the electrode/electrolyte interface has the most influence on the electrooxidation of methanol when using a semiconducting polymer electrode. It should also be noted that such charge transfer processes are also important in electrochromism and photoelectrochemical effects.44 In the first step of charge transport within PTy films, which are considered to be p-type semiconductors where holes are the majority carriers, there are two processes of importance, inter chain and intra chain transport. Intra chain transport of charge takes place along the main chain, which is facilitated due to extended conjugation. Inter chain charge transport occurs mainly across one chain to the other by a hopping mechanism.
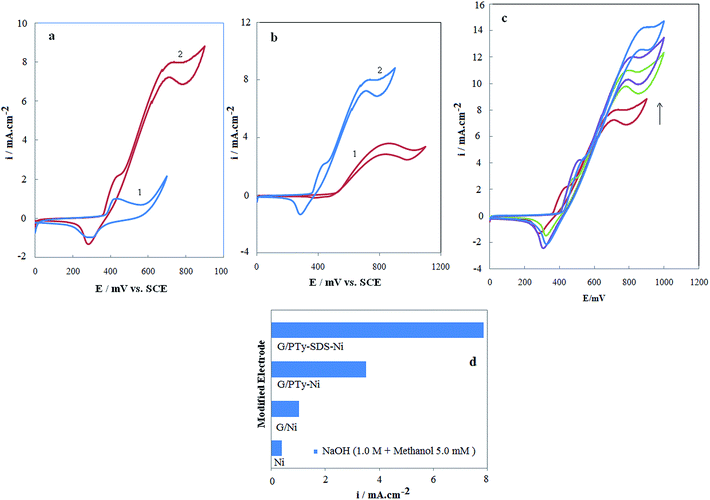 |
| | Fig. 7 (a) Cyclic voltammograms in the absence (1) and the presence (2) of 0.005 mol L−1 of methanol on the G/PTy–SDS–NiO electrode in 1.0 mol L−1 NaOH solution. The potential sweep rate was 10 mV s−1. (b) Typical cyclic voltammograms of (1) G/PTy–NiO and (2) G/PTy–SDS–NiO in 1 mol L−1 NaOH in the presence of 0.005 mol L−1 methanol, the potential sweep rate was 10 mV s−1. (c) Cyclic voltammograms of the G/PTy–SDS–NiO electrode in 1 M NaOH solution in the presence of (1) 0.005 mol L−1; (2) 0.01 mol L−1; (3) 0.015 mol L−1 and (4) 0.02 mol L−1 methanol in the solution. The potential sweep rate was 10 mV s−1, the inset shows the dependency of the anodic peak current on the concentration of methanol in solution. (d) Histograms of the current density of methanol electrooxidation on the surface of the different Ni-based modified electrodes. | |
Fig. 7b shows the cyclic voltammograms of the G/PTy–SDS–NiO and G/PTy–NiO electrodes in 1 mol L−1 NaOH solution in the presence of 0.005 mol L−1 methanol at a potential sweep rate of 10 mV s−1. As can be seen in 0.005 mol L−1 methanol the G/PTy–SDS–NiO electrode generates a higher current density and lower potential for the electrooxidation of methanol in NaOH solution. The larger methanol response at G/PTy–SDS–NiO with respect to the G/PTy–NiO electrode is proposed to be because SDS enhances the catalytic properties of nickel oxide through fine dispersion of the catalyst particles into the conductive polymer matrix which results in a drastic increase in surface area. The histograms of this behavior and the difference between the different electrode responses are presented in Fig. 7d. According to the histogram results, the presented composite (G/PTy–SDS–NiO) is a more appropriate electrocatalyst for the oxidation of methanol than others (bare Ni, G/NiO and G/PT–NiO) in alkaline solution. It is observed that the current density of the electrooxidation of methanol at the surface of G/PTy–SDS–NiO is almost constant over 300 cycles due to the stability of the electrocatalyst over this cycle number, indicating that methanol reacted with the surface and no poisoning effect on the surface was observed.
Fig. 7c shows the cyclic voltammograms of the G/PTy–SDS–NiO electrode in 1 mol L−1 NaOH solution in the presence of various concentrations of methanol at a potential sweep rate of 10 mV s−1. At the G/PTy–SDS–NiO electrode, the oxidation of methanol appeared as a typical electrocatalytic response. The anodic charge increased with respect to that observed for the modified surface in the absence of methanol and it was followed by a decrease of the cathodic charge upon increasing the concentration of methanol in solution. In the presence of 0.02 mol L−1 methanol with a potential sweep rate of 10 mV s−1, the ratio of anodic to cathodic charge in the presence of methanol was 85/6 while in its absence it was 62/54. The charge is obtained by integrating the anodic and cathodic peaks after background correction.
The anodic current in the positive sweep was proportional to the bulk concentration of methanol (Fig. 7c inset) and any increase in the concentration of methanol caused an almost proportional linear enhancement of the anodic current. So, catalytic electrooxidation of methanol on G/PTy–SDS–NiO seems to be certain. The decreased cathodic current that ensued the oxidation process in the reverse cycle indicates that the rate determining step certainly involves methanol and that it is incapable of reducing the entire high valent nickel species formed in the oxidation cycle. The electrocatalytic oxidation of methanol occurs not only in the anodic but also continues in the initial stage of the cathodic half cycle. Methanol molecules adsorbed on the surface are oxidized at higher potentials parallel to the oxidation of Ni(II) to Ni(III) species. The latter process has the consequence of decreasing the number of sites for methanol adsorption which along with the poisoning effect of the products or intermediates of the reaction tends to decrease the overall rate of methanol oxidation. Thus, the anodic current passes through a maximum as the potential is anodically swept. In the reverse half cycle, oxidation continues and its corresponding current goes through the maximum due to the regeneration of active sites for adsorption of methanol as a result of the removal of adsorbed intermediates and products. The rate of methanol oxidation as signified by the anodic current in the cathodic half cycle drops as the unfavorable cathodic potentials are approached.
Chronoamperograms were recorded by setting the working electrode potentials to the desired values and measuring the catalytic rate constant on the G/PTy–SDS–NiO electrode surface. Fig. 8a presents the chronoamperograms for the G/PTy–SDS–NiO electrode in the absence and presence of methanol over the concentration range 0.005–0.02 mol L−1. The applied potential step was 650 mV. Plotting the net currents versus the minus square root of time results in linear dependency (Fig. 8b inset). Therefore, a diffusion-controlled process is dominant for the electrooxidation of methanol. By using the slopes of these lines, we can obtain the diffusion coefficients of methanol according to the Cottrell equation [ref. 45, p. 163]:
| | |
I = nFAD1/2C*π−1/2t−1/2
| (4) |
where
D is the diffusion coefficient, and
C* is the bulk concentration. The mean values of the diffusion coefficient for methanol was 5.16 × 10
−6 cm
2 s
−1 on the G/PTy–SDS–NiO surface.
Fig. 8c shows the comparison of the chronoamperograms of the G/PTy–SDS–NiO and G/PTy–NiO electrodes. Chronoamperometry can also be used to evaluate the catalytic rate constant according to [
ref. 45, p. 503],
| |
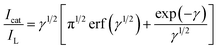 | (5) |
where
Icat and
IL are the current in the presence and absence of methanol, respectively, and
γ =
k0Ct is the argument of the error function.
k0 is the catalytic rate constant and
t is elapsed time. In cases where
γ > 1.5, erf(
γ1/2) is almost equal to unity, and
eqn (5) can be reduced to:
| |
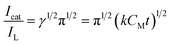 | (6) |
 |
| | Fig. 8 (a) Double step chronoamperogram of the G/PTy–SDS–NiO electrode in 1 mol L−1 NaOH solution with different concentrations of methanol: (2) 0.005 mol L−1, (3) 0.01 mol L−1, (4) 0.015 mol L−1 and (5) 0.02 mol L−1. Potential steps were 600 mV and 360 mV, respectively. (b) Chronoamperogram in 0.015 M methanol and dependency of transient current on t1/2. Fig. 8c shows compared chronoamperograms of the G/PTy–SDS–NiO and G/PTy–NiO electrodes in 0.02 mol L−1 methanol. | |
From the slopes of the Icat/IL versus t1/2 plots, the mean value of k0 obtained for methanol was 5.14 × 103 cm3 mol−1 s−1.
Electrochemical impedance spectroscopy is one of the best techniques for analyzing the properties of conducting polymer electrodes and charge transfer mechanism at the electrolyte/electrode interface. It has been broadly discussed in the literature using a variety of theoretical models.46–55 Fig. 9a shows the Nyquist diagrams of the G/PTy–SDS–NiO electrode recorded at the oxidation peak potential as dc-offset for some selected concentrations of methanol. The diagrams consist of a small semicircle terminated to depressed capacitive semicircles at the low frequency end of the spectrum. The equivalent circuit compatible with the Nyquist diagram28 recorded in the presence of methanol is depicted in the inset of Fig. 9b. To obtain a satisfactory impedance simulation of methanol electrooxidation, it is necessary to replace the capacitor C with a constant phase element (CPE) in the equivalent circuit. The most widely accepted explanation for the presence of CPE behavior and depressed semicircles on solid electrodes is microscopic roughness, causing an inhomogeneous distribution in the solution resistance as well as in the double-layer capacitance. The parallel combination of charge transfer resistance, R1, and constant phase element, CPE1, accounts for the injection of electrons from the conductive polymer to the back metallic contact. R2 and CPE2 represent methanol oxidation. To corroborate the equivalent circuit, the experimental data are fitted to the equivalent circuit and the circuit elements are obtained. Table 3 illustrates the equivalent circuit parameters for the impedance spectra of methanol oxidation with different concentrations of methanol. As can be seen from Table 3, increasing methanol concentration decreases the diameter of the semicircle, and charge transfer for methanol electrooxidation on the surface of G/PTy–SDS–NiO is lower than G/PTy–NiO. In other words, the introduction of SDS into the polymer matrix decreases the charge transfer resistance in the Nyquist plots (Fig. 9b).
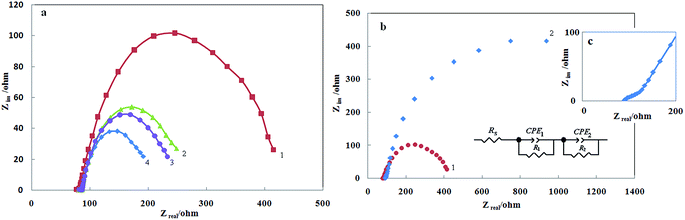 |
| | Fig. 9 (a) Nyquist diagrams of the G/PTy–SDS–NiO electrode in different concentrations of methanol in 1 mol L−1 NaOH: (1) 0.005 mol L−1, (2) 0.01 mol L−1, (3) 0.015 mol L−1, (4) and 0.02 mol L−1. DC potential is 600 mV/SCE. (b) Compared Nyquist diagrams of (1) G/PTy–SDS–NiO and (2) G/PTy–NiO electrodes in 0.002 mol L−1 methanol in 1.0 mol L−1 NaOH, (c) Nyquist plot of G/PTy–NiO in high magnification. | |
Table 3 Values of the elements in the equivalent circuit represented in Fig. 7 fitted in the Nyquist plots of Fig. 7 and the corresponding relative errors
| Rs (Ω) |
T0 × 105 (Ω−1 Sn) |
n1 |
R1 (Ω) |
T0 × 104 (Ω−1 Sn) |
n2 |
R2 (Ω) |
Cm (M) |
| 68.2 (0.36%) |
1.22 (1.12%) |
0.91 (0.52%) |
45 (0.96%) |
9.8 (0.98%) |
0.76 (0.77%) |
361 (2.35%) |
0.005 |
| 67.2 (0.25%) |
1.35 (0.86%) |
0.88 (0.23%) |
49 (0.75%) |
9.4 (2.52%) |
0.73 (0.13%) |
195 (2.15%) |
00.01 |
| 75.8 (0.18%) |
1.41 (2.14%) |
0.89 (0.64%) |
51 (0.52%) |
9.2 (1.65%) |
0.78 (0.46%) |
128 (1.85%) |
0.0.02 |
A number of mechanisms have been proposed for the electrooxidation of alcohols on Ni in alkaline solutions. Fleischmann et al.56 assumed a catalytic/intermediate role for NiOOH in the course of an anodic potential sweep.
On the basis of our study and consistent with the literature28,57–59 the following mechanism is proposed for the mediated electrooxidation of methanol on G/PTy–SDS–NiO and the corresponding kinetics are formulated. The redox transition of the nickel species present in the film is
| |
 | (7) |
and methanol is oxidized on the modified surface
via the following reaction
| |
 | (8) |
| |
 | (9) |
where Ni
3+ sites are regenerated by the power source and on the Ni
3+ oxide surface by direct electrooxidation
| |
 | (10) |
| |
 | (11) |
Eqn (8) and (9) are according to Fleischmann’s mechanism and in eqn (10) and (11) Ni3+ is used as an active surface for methanol oxidation. Observation of a new oxidation peak for methanol oxidation at a potential much more positive than that of the oxidation of Ni(OH)2 potential is in accordance with eqn (10) and (11). According to the above equation the Faradic current density can be written as
| | |
IF = (v1 + v4 + v5)F
| (12) |
In the above sequence of reactions, (7)–(9), k1 and k−1 are obviously potential dependent rate constants and are of the form:
| |
 | (13) |
| |
 | (14) |
where
ko is the chemical rate constant measured at the equilibrium potential,
α is the anodic symmetry factor and other parameters have their usual meanings. The rate laws for the reactions
(8)–(10) have the forms:
| | |
v1 = k1ΓθII − K−1ΓθIII
| (15) |
where
Γ is the total number of adsorption sites per unit area of the electrode surface,
θ represents the fractional coverage of different nickel valence states and
C* is the bulk concentration of methanol. With only the (
II) and (
III) valence states of nickel prevailing:
and the rates of change of their coverage as well as that of the intermediate compounds being:
| |
 | (18) |
| |
 | (19) |
where
Ci is the concentration of the intermediate.
Assuming that the steady state approximation dominates:
| |
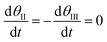 | (20) |
| |
 | (21) |
one arrives at the values of the coverage:
| |
 | (22) |
| |
 | (23) |
On the basis of this rate equation the Faradic current will be:
| |
 | (24) |
where
A is the surface area of the electrode and the corresponding charge transfer resistance is:
| |
 | (25) |
the methanol concentration dependency of
Rct are further fitted to
eqn (25) to estimate the values of the rate constants. From this equation values of the rate constants
k1 = 3.86 s
−1,
k−1 = 0.31 s
−1 and
k2 = 1.24 × 10
3 cm
3 mol
−1 s
−1 have been calculated.
6. Conclusion
PTy was formed by electropolymerization in a monomer solution containing sodium dodecyl sulfate (SDS), on a graphite (G) electrode surface. Then, Ni(II) ions were incorporated into the electrode by immersion of the polymeric modified electrode with amine groups in a Ni(II) ion solution. The morphologies and elemental components of the film were inspected by scanning electron microscopy and EDS, respectively. Theoretical calculations for Ni-polymerization were carried out at the density functional theory (DFT) level using the 6-31G(d,p) basis set for all atoms with the Gaussian 03 program package. The nickel oxide film was tested for electrooxidation of methanol in alkaline media. The modified electrodes showed electrocatalytic activity for the oxidation of methanol. A kinetic model was developed and the kinetic parameters were calculated using the methanol concentration dependency of charge transfer resistance derived from the impedance studies.
Acknowledgements
The authors would like to express their deep gratitude to the Iranian Nano Council for supporting this work.
References
- K. Scott, W. M. Taama and P. J. Argyropoulos, J. Power Sources, 1999, 79, 43–59 CrossRef CAS.
- K. Kordesch and G. Simander, Fuel Cells and their Application, VCH Verlagsgesellschaft, Weinheim, 1996, p. 151 Search PubMed.
- T. D. Jarvis, E. M. Stuve, J. Lipkowski and P. Ross, Electrocatalysis, Wiley-VCH, 1998, p. 109 Search PubMed.
- R. Parsons and T. Vander Noot, J. Electroanal. Chem., 1988, 257, 9–45 CrossRef CAS.
- K. Nishimura, K. Machida and M. Enyo, J. Electroanal. Chem., 1988, 251, 117–125 CrossRef CAS.
- E. Morallon, F. J. Cases, J. L. Vazquez and A. Aldaz, Electrochim. Acta, 1992, 37, 1883–1886 CrossRef CAS.
- P. C. Biswas, Y. Nodasaka and M. Enyo, J. Appl. Electrochem., 1996, 26(1), 30–35 CrossRef CAS.
- M. Jafarian, R. B. Moghaddam, M. G. Mahjani and F. Gobal, J. Appl. Electrochem., 2006, 36, 913–918 CrossRef CAS PubMed.
- X. Ren, P. Zelenay, S. Thomas, J. Davey and S. Gottesfeld, J. Power Sources, 2000, 86, 111–116 CrossRef CAS.
- T. Schultz, S. Zhou and K. Sundmacher, Chem. Eng. Technol., 2001, 24, 1223–1233 CrossRef CAS.
- E. Antolini, Mater. Chem. Phys., 2003, 78(3), 563–573 CrossRef CAS.
- W. D. King, J. D. Corn, O. J. Murphy, D. L. Boxall, E. A. Kenik, K. C. Kwiatkowski, S. R. Stock and C. M. Lukehart, J. Phys. Chem., 2003, 107(23), 5467–5474 CrossRef CAS.
- K. W. Park, J. H. Choi, B. K. Kwon, S. A. Lee, Y. E. Sung, H. Y. Ha, S. A. Hong, H. Kim and A. Wieckowski, J. Phys. Chem., 2002, 106(8), 1869–1877 CrossRef CAS.
- C. Fan, D. L. Piron, A. Sleb and P. Paradis, J. Electrochem. Soc., 1994, 141, 382–387 CrossRef CAS PubMed.
- I. A. Raj and K. I. Vasu, J. Appl. Electrochem., 1990, 20(1), 32–38 CrossRef CAS.
- S. Berchmans, H. Gomathi and G. P. Rao, J. Electroanal. Chem., 1995, 394, 267–270 CrossRef.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem., 1971, 31, 39–49 CrossRef CAS.
- J. Taraszewska and G. Roslonek, J. Electroanal. Chem., 1994, 364, 209–213 CrossRef CAS.
- J. R. Allen, A. Florido, S. D. Young, S. Daunert and L. G. Bachas, Electroanalysis, 1995, 7(8), 710–713 CrossRef CAS.
- A. Bettelheim, B. A. White, S. A. Raybuck and R. W. Murray, Inorg. Chem., 1987, 26(7), 1009–1017 CrossRef CAS.
- X. Tarrus, M. Montiel, E. Valles and E. Gómez, Int. J. Hydrogen Energy, 2013, 38, 12774–12785 CrossRef PubMed.
- M. U. Anu Prathap and R. Srivastava, Nano Energy, 2013, 2, 1046–1053 CrossRef CAS PubMed.
- A. Ehsani, M. G. Mahjani, M. Jafarian and A. Naeemy, Prog. Org. Coat., 2010, 69, 510–516 CrossRef CAS PubMed.
- A. Ehsani, M. G. Mahjani and M. Jafarian, Synth. Met., 2011, 161, 1760–1765 CrossRef CAS PubMed.
- X.-Y. Peng, X.-X. Liu, P.-J. Hua, D. Diamond and K.-T. Lau, J. Solid State Electrochem., 2010, 14, 1–7 CrossRef CAS.
- M. G. Mahjani, A. Ehsani and M. Jafarian, Synth. Met., 2010, 160, 1252–1259 CrossRef CAS PubMed.
- A. Ehsani, M. G. Mahjani, M. Bordbar and R. Moshrefi, Synth. Met., 2013, 165, 51–55 CrossRef CAS PubMed.
- A. Ehsani, M. G. Mahjani, M. Jafarian and A. Naeemy, Electrochim. Acta, 2012, 71, 128–133 CrossRef CAS PubMed.
- A. Ehsani, M. G. Mahjani and M. Jafarian, Synth. Met., 2012, 62, 199–204 CrossRef PubMed.
- R. Ojani, J. B. Raoof and S. Fathi, Electrochim. Acta, 2009, 54, 2190–2196 CrossRef CAS PubMed.
- F. D. Eramo, J. M. Marioli, A. A. Arevalo and L. E. Sereno, Electroanalysis, 1999, 11, 481–486 CrossRef.
- P. C. Biswas, Y. Nodasaka and M. Enyo, J. Appl. Electrochem., 1996, 26, 30–35 CrossRef CAS.
- Gauss View, Version 3.0, Gaussian Inc., Pittsburgh, PA, 2003 Search PubMed.
- M. Nasrollahzadeh, F. Babei, S. M. Sajadi and A. Ehsani, Spectrochim. Acta, Part A, 2014, 132, 423–429 CrossRef CAS PubMed.
- M. Nasrollahzadeh, A. Azarian, A. Ehsani, S. M. Sajadi and F. Babaei, Mater. Res. Bull., 2014, 55, 168–175 CrossRef CAS PubMed.
- M. Nasrollahzadeh, A. Azarian, A. Ehsani and M. Khalaj, J. Mol. Catal. A: Chem., 2014, 394, 205–210 CrossRef CAS PubMed.
- A. Ehsani, F. Babaei and M. Nasrollahzadeh, Appl. Surf. Sci., 2013, 283, 1060–1064 CrossRef CAS PubMed.
- A. Lakhtakia and R. Messier, Sculptured Thin Films, Nano Engineered Morphology and Optics, SPIE, USA, 2005 Search PubMed.
- J. H. Choi, Y. H. Lee, C. A. Kim and M. S. Jhon., J. Mater. Sci. Lett, 2000, 19, 533–535 CrossRef.
- A. Garahan, L. Pilon and J. Yin, Appl. J. Phys., 2007, 101, 014320 CrossRef PubMed.
- I. Valyukh, S. Green, H. Arwin, G. A. Niklasson, E. Wäckelgard and C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2010, 94, 724–732 CrossRef CAS PubMed.
- R. Swanepoe, J. Phys. E: Sci. Instrum., 1983, 16, 1214–1222 CrossRef.
- R. Swanepoel, J. Phys. E: Sci. Instrum., 1984, 17, 896–903 CrossRef CAS.
- C. S. C. Bose and K. Rajeshwar, J. Electroanal. Chem., 1992, 333, 235–256 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Wiley, New York, 2001 Search PubMed.
- J. Bisquert, J. Electroanal. Chem., 2001, 499, 112–120 CrossRef CAS.
- A. Ehsani, M. G. Mahjani and M. Jafarian, Turk. J. Chem., 2011, 35, 1–9 Search PubMed.
- A. Ehsani, M. G. Mahjani, M. Bordbar and S. Adeli, J. Electroanal. Chem., 2013, 710, 29–35 CrossRef CAS PubMed.
- A. Ehsani, M. G. Mahjani, R. Moshrefi, H. Mostaanzadeh and J. Shabani, RSC Adv., 2014, 4(38), 20031–20037 RSC.
- A. Ehsani, M. Nasrollahzadeh, M. G. Mahjani, R. Moshrefi and H. Mostaanzadeh, Ind. Eng. Chem., 2014, 20, 4363–4370 CrossRef CAS PubMed.
- A. Ehsani, M. G. Mahjani, M. Nasseri and M. Jafarian, Anti-Corros. Methods Mater., 2014, 61, 146–152 CrossRef CAS.
- A. Ehsani, F. Babaei and H. Mostaanzadeh, J. Braz. Chem. Soc., 2015, 26, 331–337 Search PubMed.
- A. Ehsani, M. G. Mahjani, S. Adeli and S. Moradkhani, Prog. Org. Coat., 2014, 77, 1674–1681 CrossRef CAS PubMed.
- A. Ehsani, S. Adeli, F. Babaei, H. Mostaanzadeh and M. Nasrollahzadeh, J. Electroanal. Chem., 2014, 713, 91–97 CrossRef CAS PubMed.
- A. Ehsani, Prog. Org. Coat., 2015, 78, 133–139 CrossRef CAS PubMed.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Chem. Soc., Perkin Trans. 2, 1972, 10, 1396–1402 RSC.
- M. Jafarian, M. G. Mahjani, H. Heli, F. Gobal, H. Khajehsharifi and M. H. Hamedi, Electrochim. Acta, 2003, 48, 3423–3429 CrossRef CAS.
- A. Ehsani, A. Vaziri-Rad, F. Babaei and H. Mohammad Shiri, Electrochim. Acta, 2015, 159, 140–148 CrossRef CAS PubMed.
- A. Ehsani, B. Jaleh and M. Nasrollahzadeh, J. Power Sources, 2014, 257, 300–307 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 

 is the free space wave number, λ0 is the free space wavelength, ε0 = 8.854 × 10−12 F m−1 and μ0 = 4π × 10−7 H m−1 are the permittivity and permeability of free space (vacuum), respectively and
is the free space wave number, λ0 is the free space wavelength, ε0 = 8.854 × 10−12 F m−1 and μ0 = 4π × 10−7 H m−1 are the permittivity and permeability of free space (vacuum), respectively and 

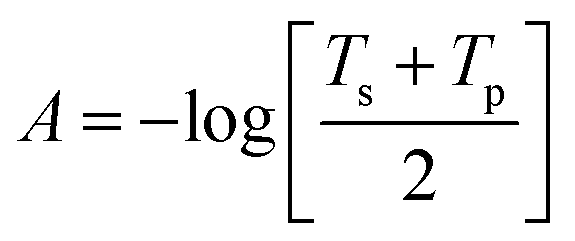 , where
, where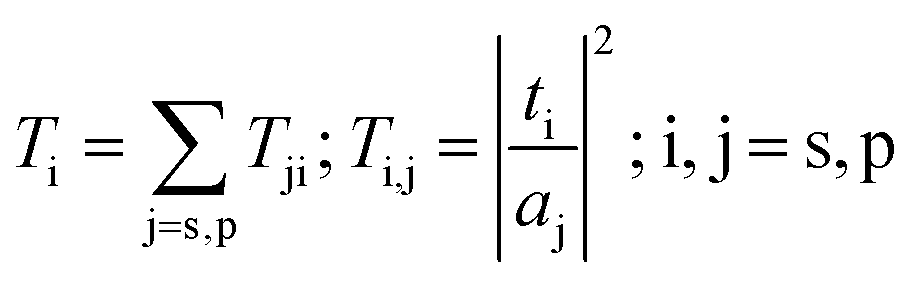 .
.


 for NiO particles embedded in PTy, where εPty and εNiO are the dielectric constants of PTy and NiO, while f is the volume fraction of dispersed particles. We have used the bulk experimental refractive indexes of NiO for homogenization.41 In our work, we extracted the refractive indexes k and n of PTy from the absorption coefficient and measured transmittance k = α
for NiO particles embedded in PTy, where εPty and εNiO are the dielectric constants of PTy and NiO, while f is the volume fraction of dispersed particles. We have used the bulk experimental refractive indexes of NiO for homogenization.41 In our work, we extracted the refractive indexes k and n of PTy from the absorption coefficient and measured transmittance k = α , where A is absorbance and hν is photon energy. It has been observed that the plot of (αhν)2 versus hν is linear over a wide range of photon energies indicating a direct type of transition. The intercept (extrapolation) of this plot (straight line) on the energy axis gives the energy band gap. The value of energy, Eg, of the composite was determined as Eg = 3.625 eV.
, where A is absorbance and hν is photon energy. It has been observed that the plot of (αhν)2 versus hν is linear over a wide range of photon energies indicating a direct type of transition. The intercept (extrapolation) of this plot (straight line) on the energy axis gives the energy band gap. The value of energy, Eg, of the composite was determined as Eg = 3.625 eV.