
Synthesis and optical properties of gold nanoparticle networks cross-linked with chain-length-controlled polymers†
Abstract
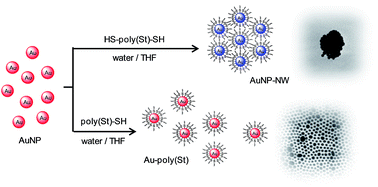
In order to control the absorption band ascribed to localized surface plasmon resonance (LSPR) of gold nanoparticles (AuNPs) in solution, we newly synthesized a gold nanoparticle network (AuNP-NW) cross-linked with chain-length-controlled polymers. The chain-length-controlled polystyrene (poly(St)) having dithiobenzoate groups at both termini was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization with a novel RAFT agent which enables the precise control of the chain length of the polymers. Both terminal groups of poly(St) were reduced from dithiobenzoate groups to thiol groups. The AuNP-NW was prepared by the self-organized cross-linking of individual AuNPs with poly(St) having thiol groups at both termini. Transmission electron microscopy images and absorption spectra of the AuNP-NW indicate the presence of a network structure. The absorption band due to LSPR of the AuNP-NW could be changed by the precise control of the distance between the AuNPs.
 Please wait while we load your content...
Please wait while we load your content...

