
Supramolecular structures ranging from nano- to macro-scale with fluorescent and organic semiconducting properties†
Abstract
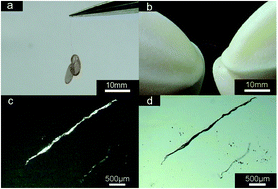
Supramolecular structures ranging from nano- to macro-scale are prepared by an ionic self-assembly (ISA) strategy with commercially available, low-cost dyes and surfactants, viz. Rhodamine 6G (R6G) and sodium bis(2-ethylhexylhexyl) sulfosuccinate (NaAOT). The organic small compounds are employed to fabricate fibers with a length up to two centimeters via ISA. They exhibit strong strength and high flexibility and could be bent without damaging the original shape. It is noteworthy that this supramolecular complex also displays both remarkable solid-state red light emission and organic semiconducting behavior. It is extremely rare that the looped nanochains comprised of organic nanoparticles are found during the formation process of the supramolecular structures. A possible mechanism for the formation of the macroscopic fibers is proposed. The meso- and macro-scale fibers can be widely used in the development of multi-scale materials with high optoelectronic performance.
 Please wait while we load your content...
Please wait while we load your content...

