
Macroporous monoliths for trace metal extraction from seawater
Abstract
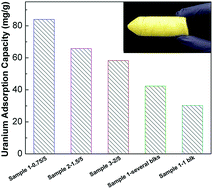
The viability of seawater-based uranium recovery depends on the uranium adsorption rate and capacity, since the concentration of uranium in the oceans is relatively low (3.3 μg L−1). An important consideration for a fast adsorption is to maximize the adsorption properties of adsorbents such as surface areas and pore structures, which can greatly improve the kinetics of uranium extraction and the adsorption capacity simultaneously. Following this consideration, macroporous monolith adsorbents were prepared from the copolymerization of acrylonitrile (AN) and N,N′-methylene-bis(acrylamide) (MBAAm) based on a cryogel method using both hydrophobic and hydrophilic monomers. The monolithic sorbents were tested with simulated seawater containing a high uranyl concentration (∼6 ppm) and the uranium adsorption results showed that the adsorption capacities are strongly influenced by the ratio of monomer to the crosslinker, i.e., the density of the amidoxime groups. The preliminary seawater testing indicates the high salinity content of seawater does not hinder the adsorption of uranium.
 Please wait while we load your content...
Please wait while we load your content...

