
Abstract
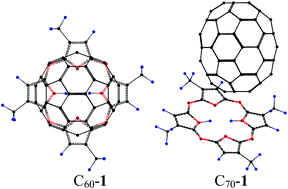
This communication reports UV-vis, fluorescence and quantum chemical investigations on the supramolecular interactions of a porphyrazine derivative, namely, 2,7,12,17-tetra-tert-butyl-5,10,15,20-tetraaza-21H,23H-porphine (1) with C60 and C70 in toluene and dichlorobenzene. A synergistic combination of rigidity in the host molecule and a shape-selective binding motif give rise to the strongest binding of C60 with porphyrazine.
 Please wait while we load your content...
Please wait while we load your content...

