
Assessment of the therapeutic potential of hesperidin and proteomic resolution of diabetes-mediated neuronal fluctuations expediting Alzheimer’s disease
Abstract
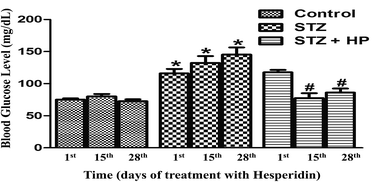
Alzheimer’s disease (AD) has been proposed as type III diabetes mellitus (DM). Prognosis and early stage diagnosis of AD is essentially required in diabetic patients to avoid extensive irreversible neuronal damage. Also, simple medication regimes including therapeutics for maintaining glucose levels and simultaneous resistance to neuronal damage are quintessential. In the present study, secretome and hippocampus proteome modulations were investigated for serum-based markers that have correlations with DM-mediated neurological alterations which extend to AD. Concurrently, the therapeutic effect of hesperidin on DM and DM-mediated neurodegeneration was investigated. Twenty one male Wistar rats were separated into three groups, namely, healthy control, diabetic (65 mg kg−1 STZ i.p., single) and diabetic administered with hesperidin (STZ i.p. + 50 mg kg−1 hesperidin orally, for four weeks). Secretome and hippocampus proteome profiling was accomplished by two dimensional electrophoresis, and proteins showing differential expression were characterized by MALDI-TOF MS PMF and validated by relative expression analysis. APO A-IV and secretory AGK were found to have prognostic and/or diagnostic potential in detecting the early stage of DM-associated AD. A novel protein, ‘WajidSaima_Diabetes protein’ (WSDP), was found to have a probable role in neural homeostasis. The therapeutic potential of hesperidin in DM and DM-mediated neuronal fluctuations has successfully been determined through proteomic resolution. Our study emphasizes the DM-mediated neuronal fluctuations that expedite AD.
 Please wait while we load your content...
Please wait while we load your content...

