
Extraction and GC-MS analysis of phthalate esters in food matrices: a review
Mario Vincenzo Russo*a,
Pasquale Avinob,
Luisa Peruginia and
Ivan Notardonatoa
aDipartimento Agricoltura, Ambiente e Alimenti, Università del Molise, via De Sanctis, 86100 Campobasso, Italy. E-mail: mvrusso@unimol.it; Fax: +39-0874-404-652; Tel: +39-0874-404-631
bDIT, INAIL Settore Ricerca, via IV Novembre 144, 00187 Rome, Italy
First published on 31st March 2015
Abstract
According to the Scopus database, using “phthalate” and “GC” as keywords, 758 papers have been found between 1990 and 2014, showing strong and increasing interest in this class of compounds from the scientific community. This review focuses attention on phthalate ester (PAE) extraction procedures, followed by GC-MS analysis, applied to food matrices and developed during the last 15 years (from 2000). In this area, 120 papers have been published, divided according to different sample preparation/extraction methods: liquid–liquid extraction (LLE), solid-phase (micro) extraction (SP(M)E), dispersive liquid–liquid (micro) extraction (DLL(M)E), headspace solid-phase micro-extraction (HS-SPME), microwave extraction, supercritical fluid extraction, ultrasonication extraction, thermal desorption extraction and Soxhlet extraction. Finally, for in-depth information, two important issues, phthalate toxicology and risk assessment and the blank problem in analytical determinations, are discussed.
Introduction
Phthalate esters (PAEs) are a group of chemicals that share basic chemical similarities and are used to make plastics more flexible and harder to break. PAEs, listed in Table 1, are chemically inert, have high density, low to medium volatility, and high solubility in organic solvents, and are easily released into the environment during aging of polymer materials. On the basis of data from several studies on phthalates concentrations in the general population, profiles of phthalates and phthalate esters varied significantly between different geographic areas. It was found that ubiquitous human exposure to phthalates occurred in both developing and industrialized countries.1 This PAE penetration in the environment and food may occur because they are not covalently bound to plastics. Therefore, they can leak into food and beverages from packaging materials and also into the environment from plastic waste.2–4 PAEs are connected with each other via hydrogen bonds and van der Waals forces. PAEs and materials retain their own chemical properties independently, so PAEs do great harm to humans if released into water, soil or the atmosphere.5,6| Phthalate | Abbreviation | CAS number | Chemical structure | Formula | MW | SIM |
|---|---|---|---|---|---|---|
| Dimethyl phthalate | DMP | 131-113 |  |
C10H10O4 | 194.18 | 163, 194 |
| Diethyl phthalate | DEP | 84-66-2 | 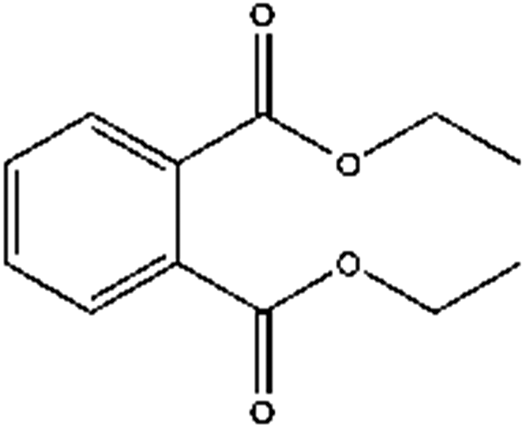 |
C12H14O4 | 222.24 | 149, 177 |
| Diallyl phthalate | DAP | 131-17-9 |  |
C14H14O4 | 246.26 | 149, 189 |
| Di-n-propyl phthalate | DPP | 131-16-8 |  |
C14H18O4 | 250.29 | 149 |
| Di-n-butyl phthalate | DBP | 84-74-2 |  |
C16H22O4 | 278.34 | 149, 205 |
| Di-isobutyl phthalate | DIBP | 84-69-5 |  |
C16H22O4 | 278.34 | 149, 223 |
| Butyl cyclohexyl phthalate | BCP | 84-64-0 | 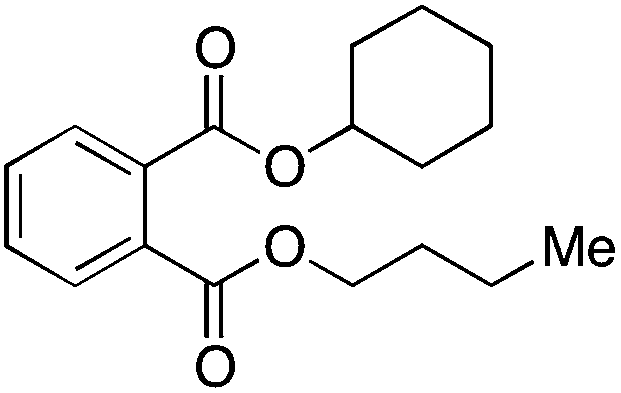 |
C18H24O4 | 304.38 | 149, 223 |
| Di-n-pentyl phthalate | DNPP | 131-18-0 |  |
C18H26O4 | 306.40 | 149, 237 |
| Dicyclohexyl phthalate | DCP | 84-61-7 |  |
C20H26O4 | 330.42 | 149, 167 |
| Butyl benzyl phthalate | BBP | 85-68-7 |  |
C19H20O4 | 312.36 | 149, 206 |
| Di-n-hexyl phthalate | DNHP | 84-75-3 |  |
C20H30O4 | 334.45 | 104, 149 |
| Di-isohexyl phthalate | DIHxP | 146-50-9 |  |
C20H30O4 | 334.45 | 149 |
| Di-isoheptyl phthalate | DIHpP | 41451-28-9 |  |
C22H34O4 | 362.50 | 149 |
| Butyl decyl phthalate | BDP | 89-19-0 |  |
C22H34O4 | 362.50 | 149 |
| Di-(2-ethylhexyl) phthalate | DEHP, DOP | 117-81-7 |  |
C24H38O4 | 390.56 | 149, 167 |
| Di-n-octyl phthalate | DNOP | 117-84-0 |  |
C24H38O4 | 390.56 | 149, 279 |
| Di-isooctyl phthalate | DIOP | 27554-26-3 |  |
C24H38O4 | 390.56 | 149 |
| n-Octyl n-decyl phthalate | ODP | 119-07-3 |  |
C26H42O4 | 418.61 | 149 |
| Di-isononyl phthalate | DINP | 28553-12-0 |  |
C26H42O4 | 418.61 | 149, 293 |
| Di-(2-propylheptyl) phthalate | DPHP | 53306-54-0 |  |
C28H46O4 | 446.66 | 149 |
| Di-isodecyl phthalate | DIDP | 26761-40-0 |  |
C28H46O4 | 446.66 | 149, 307 |
| Diundecyl phthalate | DUP | 3648-20-2 |  |
C30H50O4 | 474.72 | 149 |
| Di-isoundecyl phthalate | DIUP | 85507-79-5 |  |
C30H50O4 | 474.72 | 149 |
| Ditridecyl phthalate | DTDP | 119-06-2 |  |
C34H58O4 | 530.82 | 149 |
| Di-isotridecyl phthalate | DIUP | 68515-47-9 |  |
C34H58O4 | 530.82 | 149 |
Many consumer products contain specific members of this family, including building materials, household items, house furnishings, clothing, cosmetics, pharmaceuticals, nutritional supplements, medical devices, dentures, children's toys, glow sticks, modeling clay, food packaging, automobiles, lubricants, waxes, cleaning materials and insecticides.7–23 PAEs are also called plasticizers: for instance, di-(2-ethylhexyl) phthalate (DEHP) is one of the most widespread phthalate plasticizers, used in numerous consumer products, commodities, and building materials. Whereas in previous years DEHP was the predominant plasticizer used, with a production volume of 3–4 million tons worldwide,24 its industrial production and use have decreased in recent years. Despite only a few phthalates being produced on an industrial scale, the annual production of phthalates was estimated by the World Health Organization (WHO) to approach 8 million tons.25–27 The most important examples in this respect are DEHP, which accounts for about 50% of the world production of phthalates, di-isodecyl phthalate (DIDP), and di-isononyl phthalate (DINP). Due to their widespread application, phthalates have become ubiquitous in the environment. For example, Hubert et al. estimated the release of DEHP into the environment to be about 1.8% of the annual production.28 In addition, phthalates are stable in solution and are able to resist high temperatures.29 PAEs degrade under exposure to sunlight and are readily metabolised by aerobic microbial activity. In 2003, more than 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of phthalates were used in Western Europe: 24% DEHP and more than 50% DINP and DIDP.30 In addition, other phthalates, such as diethyl phthalate (DEP), di-n-butyl phthalate (DBP), butyl benzyl phthalate (BBP), and di-n-octyl phthalate (DnOP), are widely used.
000 tons of phthalates were used in Western Europe: 24% DEHP and more than 50% DINP and DIDP.30 In addition, other phthalates, such as diethyl phthalate (DEP), di-n-butyl phthalate (DBP), butyl benzyl phthalate (BBP), and di-n-octyl phthalate (DnOP), are widely used.
This review would like to summarize papers on PAE analysis in food matrices since 2000: extraction and GC determination will be presented and discussed in terms of analytical parameters.
Toxicology and risk assessment
Nowadays, phthalates (PAEs) represent an important problem in human health: they can accumulate in the human body, whereas chronic poisoning can cause serious damage to the liver and/or reproductive system. Humans are widely exposed to phthalates because vinyl is a ubiquitous plastic used to make anything from home furnishings (e.g., flooring, wallpaper), medical devices (e.g., catheters, blood bags), children's items (e.g., infant feeding bottles, squeeze toys, changing mats, teethers) to packaging (e.g., disposable bottles, food wrap). In addition to vinyl, humans are also exposed to phthalates in cosmetics and scented products such as perfumes, soaps, lotions and shampoos. Phthalates are also added to insecticides, adhesives, sealants and car care products. Exposure to low levels of phthalates may also arise from eating food packaged in plastic that contains phthalates or breathing dust in rooms with vinyl mini-blinds, wallpaper, or recently installed flooring that contains phthalates.31 Humans could be exposed by drinking beverages that contain phthalates, although it is not known how common this is. Phthalates are suspected to be endocrine disruptors.32,33As phthalate plasticizers are not chemically bound to PVC, they can leach, migrate or evaporate into indoor air and the atmosphere, foodstuffs, other materials, etc. Consumer products containing phthalates can result in human exposure through direct contact and use, indirectly, through leaching into other products, or via general environmental contamination. Humans are exposed through ingestion, inhalation, and dermal exposure during their whole lifetime, including intrauterine development.34
Risk assessments on phthalates have been carried out by different expert panels in Europe and America, i.e. the European Chemicals Bureau,35 European Food Safety Authority,36 European Scientific Committee on Toxicity, Ecotoxicity and the Environment,37–39 US Agency for Toxic Substances and Disease Registry,7–10 US Center for the Evaluation of Risks to Human Reproduction,11–18 US Environmental Protection Agency and International Agency on Research on Cancer (IARC).40
Phthalates exhibit low acute toxicity, with LD50 values of 1–30 g per kg body weight or even higher concentrations. Significant differences could be detected in different species and between males and females. All phthalates have tested negative for mutagenicity and/or genotoxicity, whereas with regard to carcinogenicity19 the activity of DEP is questionable, for DINP no hints of carcinogenicity were obtained, DBP seems to be associated with tumor-promoting activity, and exposure to DEHP produced hepatocellular carcinoma in rodents, along with a variety of other hepatocellular effects.41 Furthermore, the European Food Safety Authority established acceptable daily intakes of certain PAEs (DBP 0.01; BBP 0.5; DEHP 0.05; DNP 0.15; DDP 0.15 mg per kg body weight per day).42 Therefore, IARC recently evaluated DEHP and changed its classification from “possibly carcinogenic to humans” to “not classifiable as to carcinogenicity in humans”.40 To calculate an adult individual's daily integrated exposure to different phthalate esters via different exposure media, the following equation was used:43,44

However, in recent years toxicological concerns have arisen regarding the possible endocrine-disrupting potential of phthalates.45,46 Because of the lack of human data, studies with experimental animals were evaluated and the effects (no observed adverse-effect level, NOAEL, and lowest observed adverse-effect level, LOAEL) were compared to available human exposure data and estimations, respectively. Based on these data, concerns were expressed that human exposure to phthalates may result in reduced sperm counts, histological changes in testes, and reduced fertility.19 Finally, the presence of PAEs can interfere with the secretion of human hormones and can cause disorders of the human secretory system. Accumulation of PAEs over a long period can lead to deformity or cancer in humans.47
Owing to the lack of knowledge on the human health effects of phthalates, several international government agencies (e.g., the Food and Drug Administration and the National Institute of Environmental Health Sciences) have studied these compounds. For instance, di-(2-ethylhexyl) phthalate (DEHP) is listed as “reasonably anticipated to be a human carcinogen” in a report on carcinogens published by the US National Toxicology Program. Basically, the current levels of seven phthalates, i.e. dimethyl phthalate (DMP), diethyl phthalate (DEP), di-isobutyl phthalate (DiBP), dibutyl phthalate (DBP), butyl benzyl phthalate (BBP), isobutyl cyclohexyl phthalate (iBcEP) and di-(2-ethylhexyl) phthalate (DEHP), posed “minimal” concern for causing reproductive effects. It is reasonably recognized that high DBP levels may adversely affect human reproduction or development, whereas high levels of exposure to DEHP through the use of medical tubing and other plastic devices for feeding, medicating, and assisting the breathing of newborn infants may affect the development of the male reproductive system. The European Union, United States and several other countries have begun to regulate PAE exposure; on the other hand, the maximum allowable limit for any PAE is 1 ppm in China and Taiwan.
Foods are the major source of human exposure to phthalates, so it is important to monitor the levels of phthalates in various foods to provide data for human exposure assessment. Since phthalates are ubiquitous environmental contaminants, contamination of the environment is one of the sources of the phthalates present in foods at various levels.48 Although the use of phthalates in food packaging materials is decreasing, there are still many products that contain phthalates and adipates as plasticizers and are used for food packaging and processing. Migration of plasticizers from these products is the major path of phthalates and adipates into foods that are in contact with these products.
Phthalate residues in foods and beverages are regulated internationally: in many countries, PAEs are clearly prohibited, as non-food substances, for use in food. In Europe, for example, the use of phthalates in materials destined to be in contact with foodstuffs is subject to Commission Regulation 10/2011 of 14 January 2011. This regulation pays special attention to certain PAEs and requires their complete prohibition as of 1 January 2015: these are BBP, DBP and DEHP. There have been reports from the US Food and Drug Administration that certain foods and beverages, particularly fruit juices, contain high levels of phthalates.49 In some cases, deliberate adulteration of soft drinks with phthalate esters has been reported.
Food might be contaminated via migration from packaging materials, via different kinds of environmental sources, or during processing. For instance, alcoholic drinks in plastic containers are a particular risk, since the bevearage containing ethanol provides a very good solubility for PAEs and is leaching the PAEs into the beverages from the plastic contact materials. In a previous paper, the same authors reported how the contamination risk increases with liquors having high ethanol content.50
There is no acceptable concentration in wines and spirits. At the present time, and at least until the beginning of the ban, the EU regulation 10/2011 of 14 January 2011 is applicable: it is therefore necessary to monitor the presence of these molecules, especially those listed as most toxic (BBP, DBP and DEHP) in wines, spirits and soft drinks.
Finally, because DEHP comprises a quarter of all plasticizers ever produced,51,52 we focus more attention on it. The environmental impact of DEHP is very strong: its extremely high hydrophobicity (its calculated logKow is 8.6) makes it highly persistent and bioaccumulating.53–58 The potential toxicity of DEHP and its metabolites has caused great alarm, especially for high-risk groups such as children and patients on hemodialysis or in critical care units.59,60 DEHP has proved to have carcinogenic properties,61,62 as it acts as a hepatic peroxisome proliferator in rodents, increasing both the size and number of their hepatic peroxisomes, which leads to increased cellular oxidative stress and subsequent oxidative genome damage.63 Recently, new light has been shed on the DEHP action mechanism: its primary metabolic by-product, mono-(2-ethylhexyl) phthalate (MEHP), exhibited good affinity for retinoic acid receptors in several human tissues,64 and retinoids are well-known teratogenic agents. As no affinity for retinoic acid receptors has been observed for DEHP, a detailed description of the kinetics of the metabolic conversion of DEHP to MEHP is essential.
Analytical procedures
According to a search performed on the scientific database Scopus, using “phthalate” and “GC” as keywords, 758 papers were found between 1990 and 2014. In particular, during the 1990s only 70 papers appeared, whereas there were 66 publications in 2014: this gives evidence of strong and increasing interest in this class of compounds. In this review, we focus our attention on PAE extraction procedures, followed by GC-MS analysis, applied to food matrices and developed during the last 15 years (from 2000). This restricts the survey to about 120 papers, divided according to different sample preparation/extraction methods: liquid–liquid extraction (LLE), solid-phase (micro) extraction (SP(M)E), dispersive liquid–liquid (micro) extraction (DLL(M)E), microwave extraction, supercritical fluid extraction, ultrasonication extraction, thermal desorption extraction and Soxhlet extraction. In general, almost 90% of these papers employ LLE, SPE/SPME, or DLLE methods (including their variations). All these analytical extraction procedures are followed by gas chromatography coupled with mass spectrometry (GC-MS).An important analytical issue: the blank problem
Before beginning to describe the different analytical procedures used for analyzing PAEs in food matrices, there is a big issue regarding sample contamination by DEHP and DBP during the analysis, i.e. the cause of the blank problem. In fact, the ubiquity of some phthalates causes severe problems in the determination of their content in food. Special measures are required to keep the background level low.65 Furthermore, it should be noted that PAEs present as vapors or part of the particulate matter in air are the main source: they contaminate all surfaces, particularly glassware, plastic articles and our skin. On the other hand, blank values are defined as “a reading or result originating from the matrix, reagents and any residual bias in the measurement device or process, which contributes to the value obtained for the quantity in the analytical procedure”.66 Phthalates, in particular di-(2-ethylhexyl) phthalate (DEHP) and dibutyl phthalate (DBP), often cause “blank” problems when analyzed at low concentrations. Substantial peaks are also obtained without a sample, and analytical results tend to be too high because of phthalates introduced into the sample during analysis.67 Furthermore, the widespread presence of PAEs in the laboratory environment, including air, glassware and reagents, can produce false positive results.68 It is common practice to heat the glassware used for sample preparation and redistill solvents. Even laboratories free of materials containing phthalates, such as PVC flooring and electric cables, have been built. For the same reason, there are no foods free of DEHP and DBP. Other phthalates, such as di-isononyl, di-isodecyl or benzyl butyl phthalate, cause fewer blank problems because of lower environmental background levels and higher detection limits required (higher legal limits). Blank problems may lead to a difficult start if several sources contribute to system contamination. This could cause positive/negative artifacts. To avoid PAE contamination, some authors describe a method for cleaning all the glassware used: soaking in acetone for at least 30 min, then washing with acetone, rinsing with hexane, and drying at 120 °C for at least 4 h. Before using, all the glassware and reagents are checked for potentially occurring phthalate contamination (e.g., hexane and dichloromethane by GC-MS analysis).69,70 Other authors couple this procedure with the use of solvents at pesticide grade (e.g., dichloromethane, chloroform, tetrachloroethylene, carbon tetrachloride) and absolute ethanol; furthermore, acetone, n-hexane, dichloromethane and carbon disulfide are also distilled before use.50,71–73 Fankhauser-Noti and Grob67 confirmed that if the use of phthalates-containing plastic materials, such as PVC, is avoided during sample preparation, blank values result mainly from input via air and particulates; special design of laboratories, i.e. omitting the use of PVC for coating the floor, construction of cable ducts, etc., could minimize indoor air contamination by PAEs and hence reduce the blank values of certain PAEs. Simultaneously, an interesting solution is proposed. The paper shows the sample contamination from various sources quantified and related to DBP and DEHP in the laboratory air. The injection blank is tested by injecting hexane from an auto-sampler vial containing aluminum oxide: the removal of phthalates by aluminum oxide proves efficient for non-polar solvents (pentane and hexane). On the other hand, the authors do not test this for more polar solvents, because it is expected that strong solvents would extract phthalates from aluminum oxide rather than being purified.Another important contamination source is the outer wall of the syringe needle, due to PAEs absorbed from the laboratory air.74 Cleaning the outer needle wall is the simplest solution, but this would require the immersion of a needle section of about 5 cm in a cleaning solvent (such as hexane containing activated aluminum oxide or silica gel for PAE removal). The problem arises when an auto-sampler is used. The authors propose two approaches for minimizing PAE transfer from the outer needle wall into the column: (i) minimizing desorption from the needle wall by fast injection into a cool chamber (40 °C) and (ii) removal of phthalates evaporated from the needle wall through the split outlet while the pre-column is in back-flush mode. Both allowed blanks to be obtained in the order of 0.1 pg for DIBP and DBP and 1 pg for DEHP.
Solid-phase (micro) extraction method
Solid-phase extraction (SPE) is an increasingly useful sample preparation technique. With SPE, many of the problems associated with liquid–liquid extraction (see next section) can be prevented, such as incomplete phase separations, less-than-quantitative recoveries, use of expensive, breakable specialty glassware, and disposal of large quantities of organic solvents. SPE is more efficient than liquid–liquid extraction, allows quantitative extractions that are easy to perform, is rapid, and can be automated. Solvent use and lab time are reduced.SPE is most often used to prepare liquid samples and extract semi-volatile or nonvolatile analytes, but can also be used with solids that are pre-extracted into solvents. SPE products are excellent for sample extraction, concentration, and cleanup. They are available in a wide variety of chemistries, adsorbents, and sizes. Selecting the most suitable product for each application and sample is important.
Because of its versatility, SPE can be used in different applications and could be modified. Firstly, we would like to start from basic SPE. Table 2 summarizes all the main analytical parameters reported in different SPE methods.
| Phthalate | Sorbent/elution solvent | Matrix | r2 | Repeatability | LOD/LOQ | Recovery | Ref. |
|---|---|---|---|---|---|---|---|
| DMP, DEP, DBP, BBP, DiBP, DEHP | C18/dichloromethane + methanol | Red wine and white wine | 0.990–0.997 | 13–21 | 15–18/24–29a | 58–92 | 70 |
| DBP, BBP, DEHP | PLRP-S/ethyl acetate + methanol | Bottled water and tap water | >0.997 | 8–15 | 0.0001–0.02/—a | 50–65 | 75 |
| DMP, DEP, DBP, BBP, DEHP, DNOP | Oasis MAX/acetonitrile | Ham sausage | 0.9914–0.9944 | — | 0.16–0.61/0.53–2.01b | 87.3–108.0 | 77 |
| DEP, DAP, DiPP, DBP, BBP, DEHP, DHP, DCHP, DPP, DOP, DDP | MgSO4 + PSA/acetonitrile + hexane (drops) | Vegetable oils | 0.9979–0.9997 | 4.7–10.9 | 3–32/7–75b | 68.2–95.5 | 78 |
| DMP, DEP, DBP, BBP, DEHP | Florisil/dichloromethane + hexane | Mussel | 0.9910–0.9991 | 7–10 | 400–2600/—c | 81–115 | 82 |
| DMP, DBP, BBP, DEHA, DEHP | Oasis HLB/dichloromethane + hexane (1 + 1) and dichloromethane + acetone (1 + 1) | Bottled water | — | — | 0.010–0.460/—a | 93–125 | 83 |
| 18 PAEs | Silica-PAS/acetonitrile | Edible vegetable oils | 0.9917–0.9995 | 5.4–12.6 | 10–100/—b | 64.6–104.6 | 84 |
| DMP, DEP, DBP, BcEP, BBP, DEHP | Carbograph 1/CS2 | White and red wine | 0.9992–0.9997 | 0.9–7.8 | 0.2–14/0.5–25a | 78–105 | 71 |
| DMP, DEP, DBP, DiBP, BcEP, BBP, DEHP | XAD-2/carbon disulfide | Light alcohol and soft drinks | 0.9119–0.9976 | 3.6–6.3 | 0.2–20/0.6–41a | 88–102 | 85 |
| DMP, DEP, DBP, BcEP, BBP, DEHP | XAD-2/ethyl acetate + acetone | Wines packed in glass bottles and Tetrapak brick, and vodka liqueur | 0.9906–0.9947 | 7.7–9.4 | 1.21–2.51/2.42–3.78a | 91.6–103.8 | 50 |
| DMP, DEP, DPP, DBP, BBP, DEHP, DOP | C18 or florisil/hexane + acetone mixture | Sport drink, tea, coffee, fruit juices | 0.996–0.998 | 9–15 | 3–4/10–10a | 84–105 | 86 |
| DMP, DEP, DBP, DEHP, DNOP | PSA/acetonitrile or formic acid + methanol (3 + 97) | Seafood, fish, mollusk and prawn | 0.9984–0.9998 | 4–21 | 0.015–0.048/— | 57–119 | 87 |
| 16 PAEs | Magnetic carbon nanotubes/acetone | Juice drink, mineral water, tap water | 0.9821–0.9993 | 5.0–14.6 | 0.003–0.06/0.010–0.13a | 74.2–125.6 | 88 |
| DMP, DEP, DiBP, DBP, BBP, DEHP, DNOP | Magnetic graphene/ethyl acetate | Water | 0.9973–0.9991 | 5.1–8.4 | 0.010–0.056/0.035–0.19a | 88–110 | 89 |
One of the first papers present in the literature where SPE for determining PAEs was applied to a food matrix is dated 2002: Brossa et al.75 used PLRP-S as sorbent and ethyl acetate as elution solvent. A PLRP-S phase is made of rigid macroporous spherical particles of polystyrene and poly(divinyl benzene). They are physically and chemically stable across the complete pH range. The authors managed to set up an on-line system for analyzing three PAEs, along with 112 other endocrine disruptors, in bottled and tap water samples by pre-concentrating only 15 mL of the sample, and simultaneously identified the analytes in a 55 min single run. Del Carlo et al.70 developed a SPE method for determining six PAEs in red and white wines. In particular, the paper shows a SPE procedure modified from EPA method 506,76 which reports PAEs determination in drinking water. The authors optimized the procedure in terms of C18 phase amount, phase conditioning, sample treatment, and sample size. The SPE procedure used for wine samples analysis is as follows: 2.5 g C18 phase conditioned with 10 mL dichloromethane, plus 2.5 mL methanol; then 5 mL sample, diluted to 50 mL with water plus 2 g mL−1 NaCl, was loaded at a 1 mL min−1 flow rate. Finally, the paper also shows a statistical evaluation of the total and individual phthalate concentrations in white and red wines; this allows confirmation that the total PAEs level is not affected by the vinification process, and neither are the DBP, BBP and DEHP contents. In 2010 Guo et al.77 applied the SPE method to ham sausages. A ham sausage is a very complicated sample matrix, containing fat, proteins, carbohydrates, vitamins, cholesterol and inorganic salts. In addition, a packaging film for direct food contact can also contain different additives, including PAEs. Therefore, to extract PAEs from the sausage matrix with as little interference and as much recovery as possible is a most difficult and critical process. Among the different sorbents tried, the authors used a Waters Oasis MAX cartridge, obtaining good recoveries. In fact, because the MAX cartridge possesses a macroporous copolymer sorbent [poly(divinyl benzene-co-N-vinylpyrrolidone)] bonded to strong anion-exchange quaternary amine groups, and the ester groups of phthalates are electronegative, electrostatic attraction between the quaternary amine and ester groups will favorably retain phthalates on the MAX cartridge. Six phthalates, i.e. DMP, DEP, DBP, BBP, DEHP and DOP, were well separated in less than 14 min without significant interference from the sample matrix. Furthermore, the paper gives evidence of an important issue regarding migration: the DEHP concentrations decrease progressively with depth beneath the ham sausage surface and this phthalate can be detected at a depth of 10 mm after enough storage time. Four Italian researchers set up a SPE method for determining 11 PAEs:MgSO4 and primary-secondary amine (PSA) sorbents were used, along with acetonitrile (and some drops of hexane).78 The PSA sorbent has been shown to be very effective in cleanup of food residues, removing the greatest number of food matrix interferences.79–81 PSA, alone or with graphite carbon, has been considered an interesting agent in SPE for cleanup of pesticide residues in analysis of fruits and vegetables.
An interesting paper published by Sánchez-Avila et al. concerns the development of a multi-residue method for the determination of organic micropollutants in mussels using GC-tandem MS.82 Actually, the authors developed a method for analyzing sixteen polycyclic aromatic hydrocarbons (PAHs), five PAEs, seven polychlorinated biphenyls (PCBs), six polybrominated diphenyl ethers (PBDEs), six alkyl phenols (APs), three organochlorine pesticides and their isomers or degradation products (OCPs) and bisphenol A in seawater, river water, wastewater treatment plant (WWTP) effluents, sediments and mussels using Florisil as sorbent and dichloromethane/hexane as extraction solvent. Lacorte et al. published another paper83 where Oasis HLB was used as sorbent and dichloromethane–hexane (1 + 1) and dichloromethane–acetone (1 + 1) as extraction solvents. In particular, Oasis HLB sorbent is a universal polymeric reversed-phase sorbent developed for the extraction of a wide range of acidic, basic, and neutral compounds from various matrices using a simple, generic protocol. Since Oasis HLB sorbent is water-wettable, it retains its capability for higher retention and excellent recoveries even if the sorbent runs dry, which means there is no need to take extraordinary steps to keep the sorbent beds from drying out during the critical stages prior to sample loading.
A method for determining 18 PAEs in edible vegetable oils using silica-PAS as sorbent has been developed by Liu et al.,84 whereas Russo et al. developed two SPE methods based on Carbograph 1 for testing for PAEs in wine samples71 and on XAD-2 for analyzing PAEs in light alcoholic drinks (alcohol content below 6% alcohol by volume) and soft drinks,85 in both white and red wines packed in glass bottles and Tetrapak bricks, and in vodka liqueur.50 In particular, Russo et al. investigated the breakthrough curves of six PAEs: retention of compounds with aliphatic substituents increases with the number of carbon atoms in the aliphatic chain (DMP, DEP, DBP), whereas compounds with different substituents (DEHP, BcEP and BBP) are retained less. Furthermore, comparing the breakthrough volumes obtained with XAD-2 as adsorbent with those found using Carbograph 1, the difference in retention among PAEs with different lengths of aliphatic chain is less marked. In addition, the authors investigated whether the presence of NaCl increased PAE adsorption by reducing the solubility of these compounds whereas acetone, as co-elution solvent with ethyl acetate, being more wettable and miscible in both hydrophobic and hydrophilic solvents, removes water from the gel phase of the adsorbent. In this way, better and more efficient penetration of ethyl acetate into the XAD-2 microspheres allows good PAE recoveries and LOQs to be obtained.
Wu et al. investigated 7 PAEs in sport drinks, tea, coffee and fruit juices.86 The method used was based on two different SPE procedures performed using C18 and Florisil with a hexane/acetone mixture as elution solvent. The two columns were used due to the different particle sizes present in the samples: specifically, the C18 column was selected for samples with particle size less than 0.45 μm (sport drinks, tea, coffee), whereas the Florisil column was used for samples with particle size ≥ 0.45 μm (fruit juices).
Finally, a sensitive and reliable multi-residue method for the determination of trace amounts of endocrine-disrupting chemicals, including five PAEs, five monoalkyl phthalate esters (MPEs), four alkyl phenols (APs) and bisphenol A (BPA), in seafood was developed in 2014.87 The pretreatment method reduces blank contamination by PAEs, with background levels between 0 and 8.18 ng g−1, and obtains low matrix effects for BPA.
Two interesting papers use magnetic SPE (MSPE) methods: Luo et al. employ88 magnetic carbon nanotubes (CNTs) for fast extraction from beverages, whereas Ye et al.89 employ magnetic graphene composites synthesized via a simple hydrothermal reaction for analyzing PAEs in water samples. In MSPE, the separation process can be performed directly on crude samples containing suspended solid materials without the need for additional centrifugation or filtration, which makes the separation easier and faster. Their strong adsorption property makes magnetic CNTs an excellent candidate for serving as adsorbent in MSPE. On the other hand, the magnetic graphene composites used as adsorbent for PAEs analysis have several advantages, including high adsorption capacity and good extraction ability.
Solid-phase micro-extraction (SPME) is a pre-concentration technique which has recently been introduced for the extraction of organic compounds.90–92 Before 2000 only a few papers were dedicated to PAE determination using SPME:Möder et al.93 extracted some phthalate esters in a study on the characterization of water-soluble compounds in slurries by a SPME-HPLC-MS method, and Kelly and Larroque94 used SPME-HPLC-UV to determine diethyl phthalate ester in water samples. These studies used polydimethylsiloxane–divinyl benzene (PDMS–DVB)94 and Carbowax-coated93 fibers. The advantages of this method are simplicity, no use of solvents, sensitivity, and portability; it is also relatively independent of the design of the instrument that is used subsequently. As the fiber coating plays a key role in SPME, the development of fiber coatings for highly efficient extraction of the analytes of interest is an important research direction in SPME. Table 3 shows the main analytical parameters for this method.
| Phthalate | Sorbent/time | Matrix | r2 | Repeatability | LOD/LOQ | Recovery | Ref. |
|---|---|---|---|---|---|---|---|
| DMP, DEP, DBP, BBP, DEHP, DOP, DNP | CW–DVB/40 min | Drinking water | 0.8438–0.999 | 15–21 | 5–20/— | — | 95 |
| DMP, DEP, DBP, BBP, DEHP, DOP | PDMS–DVB/20 min | Bottled water | 0.9980–0.9994 | 3.4–16 | 2–103/— | — | 96 |
| DMP, DEP, DBP, DAP, DNOP | Molecularly imprinted polymer/30 min | Bottled water, tap water | 0.9898–0.9993 | 3.08–7.81 | 3–21/20–34 | 94.54–105.34 | 97 |
| DPP, DBP, DiBP, BBP | MWCNTs–PPy/25 min | Mineral water, tap water | 0.9891–0.9959 | 8.4–10.5 | 50–100/— | 96–105 | 101 |
| DMP, DEP, DBP, BBP, DEHP, DINP, DOP | PDMS–PA/20 min | Water | >0.9917 | <15 | 2–8/7–27 | — | 102 |
| DMP, DEP, DiBP, DBP, BBP, DEHP, DNOP | Polypyrrole-coated Fe3O4/15 min | Tap water | 0.9895–0.9910 | 3.4–11.7 | 6–68/20–30 | 91.1–113.4 | 103 |
| DMP, DEP, DiPP, DPP, DBP, DHP, BBP, DCHP, DEHP, DOP, DBzP | PDMS–DVB/30 min | Mineral water | — | — | 0.7–1.6 | — | 104 |
| DBP, DiBP, BBP, DEHP | TiO2 nanoparticles/20 min | Bottled water | >0.994 | 11.2–15.2 | 50–120/170–400 | — | 105 |
| DMP, DiPP, DEP, DPP, DiBP, DBP, DiAP, DEHP, DOP, BBP | PA + PDMS/20 min | Bottled water, tap water | 0.9613–0.9987 | 11.3–18.5 | 10–60/40–190 | 73.9–100.7 | 108 |
| DBP, BBP | Polyacrylate fiber/30 min | Food simulant | 0.9854–0.9905 | 11.7–16.2 | 80–310/200–500 | — | 109 |
The first paper using SPME followed by GC-MS for analyzing PAEs in drinking water samples is dated 2001:95 the authors studied different non-polar and polar fibers (PDMS, PA, PDMS–DVB, DVB–Carboxen–PDMS, CW–DVB). On the other hand, in 2005 Polo et al.96 investigated different commercial fibers, polydimethylsiloxane (PDMS), polydimethylsiloxane–divinyl benzene (PDMS–DVB), polyacrylate (PA), Carboxen–polydimethylsiloxane (CAR–PDMS) and carbowax–divinyl benzene (CW–DVB), as well as the extraction mode, exposing the fiber directly to the sample (DSPME) or into the headspace over the sample (HS-SPME), and different extraction temperatures. The optimized micro-extraction showed a linear response and good precision for all target analytes. Detection limits were estimated considering the contamination problems associated with phthalate analysis.
He et al. developed a novel method based on a molecularly imprinted polymer (MIP) that was applied to a SPME device followed by GC-MS.97 Molecularly imprinted polymers (MIPs), a tool used to prepare molecular recognition materials, have shown potential applications as the stationary phase in chromatography and as materials for solid-phase extraction,98–100 simulated enzyme catalysts, chemical sensors, membrane separation, and many other fields. They are quite suitable as a material for SPME fibers due to their high selectivity, chemical stability, and easy preparation. Two interesting papers deal with direct immersion SPME (DI-SPME) followed by GC-MS.101,102 Both groups of authors optimize the parameters affecting extraction efficiency, including the type of fiber, desorption temperature, desorption time, adsorption time, and sample matrix. In Asadollahzadeh et al.'s paper, the electrodeposited coating exhibits a porous structure with a high specific surface area and adsorption capacity and therefore high PAE extraction efficiency. The composite coating is a solid porous sorbent, and extraction occurs on the surface of the pores. This may be due to the fact that both multi-walled carbon nanotubes (MWCNTs) and a polypyrrole composite (PPy) take part in the PAE extraction. The coating also has a long lifetime, with excellent adhesion onto a steel surface. The lifetime of the coating is such that a single fiber could be used at least 60 times for direct immersion SPME analysis of phthalate esters in water. In Lee et al.'s paper, commercially available polydimethylsiloxane (PDMS) and polyacrylate (PA) housed in a manual holder were tested. An interesting issue regards the role of NaCl. The peak areas for DMP and DEP increase with a rise in NaCl concentration, but those for other esters decrease at a NaCl concentration higher than 0.06 g mL−1 instead. This difference shows that, with an increase in NaCl concentration, the more challenging PAEs with higher molecular weight could be extracted. NaCl increases the partitioning of DMP, DEP, and BBP from water into the headspace, whereas the partitioning of heavier PAEs is suppressed when the concentration of NaCl is above 10%. Meng et al.103 synthesized polypyrrole (PPy)-coated Fe3O4 magnetic microspheres and applied them as a magnetic sorbent to extract and concentrate phthalates from water samples. The PPy-coated Fe3O4 magnetic microspheres had the advantages of a large surface area and convenient and fast separation ability. The PPy coating on the magnetic microspheres contributed to PAE pre-concentration from water samples, due to π–π bonding between the PPy coating and the analytes. In addition, the coating could prevent aggregation of the microspheres, and improve their dispersibility. This fast, simple and convenient method for analyzing PAEs in water samples has great potential in applications for detecting other compounds such as polycyclic aromatic hydrocarbons (PAHs) in water. An ultra-trace SPME-GC-MS method has been developed for investigating the potential presence of a broad range of organic compounds, such as hormones, alkyl phenols, bisphenol A and PAEs, as well as pharmaceutical substances, in two French brands of bottled natural mineral waters.104 This is the first time that a wide range of hormones and pharmaceutical substances have been tested for in natural mineral waters. The results obtained in this study underline the complexity of devising a reliable measure to quantify the contamination of a sample at ultra-trace levels. The authors used analytical procedures involving glassware, equipment, hoods, and rooms specifically dedicated to trace analysis, allowing ng L−1 concentrations to be detected by lowering background laboratory contamination to the minimum achievable. However, the authors themselves state that the blank levels, although very low, could probably be further lowered if manipulations were performed in a clean room or by using glove boxes under a controlled atmosphere.
Another DI-SPME method involves a novel fiber prepared by electrophoretic deposition of titanium dioxide nanoparticles (nano-TiO2) on a stainless steel wire.105 The effects of various parameters on the efficiency of the SPME process, such as the mode of extraction, extraction temperature, film thickness of the SPME fiber, salt content, extraction time and stirring rate, were investigated: under optimized conditions, the LODs for PAEs vary between 0.05 and 0.12 μg L−1. Despite the potential applications of TiO2 as a sorbent, its use as a SPME coating is hindered by the difficulties involved in its immobilization on SPME fibers, and studies on this subject have been scarce. The preparation of a TiO2 coating for SPME applications can be accomplished using adhesives such as epoxy resin. Electrophoretic deposition (EPD) is a cheap, simple and useful colloidal processing technique, in which charged particles can be deposited on an oppositely charged electrode surface from a stable suspension of the particles dispersed in a suspending liquid, under the influence of an applied DC electric field. EPD is able to produce uniform deposits with high microstructural homogeneity, to provide adequate control of deposit thickness, and to deposit coatings on a wide range of shapes, 3D complexes and porous structures.106 The major advantages and attractiveness of EPD are its simplicity, high rate of deposition, few restrictions regarding the shape of the substrate, reliability of the process, the possibility of room temperature processing, suitability for co-deposition of various materials and no requirement for binder burnout, as the green coating contains few or no organics. EPD offers easy control of the thickness and morphology of a deposited film via simple adjustment of the operating conditions, including the deposition time, applied potential and concentration of the suspension.107 The coating has a porous structure, with a high specific surface area and adsorption capacity and therefore high extraction capacity and efficiency for phthalate esters. Finally, the coating also has a long lifetime: a single fiber could be used up to 60 times for DI-SPME analysis of PAEs in water.
Finally, in 2014 two papers were published regarding PAE determination in drinking water108 and a food simulant.109 The use of a DI-SPME ionic liquid (IL) followed by GC-MS, due to the high boiling points and water solubility of phthalates, has proven to be a suitable analytical technique for the analysis of phthalates at low concentrations and a large number of samples. In addition, the paper shows that the PAE concentrations measured in Portuguese bottled waters do not represent any risk to adult health, nor do the tap water samples showing PAE concentrations below 6 μg L−1, the maximum admissible concentration in water established by the US Environmental Protection Agency. On the other hand, Moreira et al. determined PAE content using SPME cooled with liquid nitrogen.109 Ultrapure water is used as a simulant for liquid foods. The use of cold SPME has allowed the quantification of PAEs that are found in low concentrations in samples, because of increased efficiency in the extraction step. Cold SPME can be used for various compounds and is a technique that is potentially applicable for the study of endocrine disruptor compounds (EDC) and other organic compounds that are present in trace concentrations in different matrices.
In SPME, the extraction of target analytes from the sample matrix to the fiber is conducted either directly, with the coated fiber directly immersed in the liquid sample (direct SPME), or in the headspace (HS-SPME), where the extracting fiber is suspended above the sample. Although suitable for the analysis of phthalate esters in water, sediments and sludge, the direct SPME method cannot be applied to complex matrices such as milk and other dairy products, which often contain fat and other biological components. The advantage of the HS approach is that it eliminates co-extraction of undissolved particles and non-volatile components in the sample matrix. Therefore, HS-SPME is a potentially useful tool for selectively extracting PAEs, as the fiber is not directly submerged in the matrix. During the last decade, different papers have been published using HS-SPME followed by GC-MS (Table 4). The first two publications relate to PAE determination in cow milk110 and human milk samples,111 whereas Carrillo et al.112 investigated PAEs in wine, comparing three different fiber compositions, i.e. carbowax–divinyl benzene (CW–DVB), polyacrylate (PA) and polydimethylsiloxane–divinyl benzene (PDMS–DVB). For these fibers, the authors investigated the influence of the extraction temperature, salting-out effect and sample volume, showing that: (a) high temperatures favor extraction; (b) the optimal sample volume decreases with increasing fiber polarity; and (c) the optimal value of salt concentration increases with increasing fiber polarity. PA fiber was discarded because it displayed poor linearity and repeatability for DEHP. CW-DVB and PDMS-DVB fibers can be successfully used for extracting phthalates from wine. The repeatability values obtained with both fibers are similar, but the former is more efficient for extracting lower-molecular-weight PAEs while the latter is better for higher-molecular-weight PAEs. In addition, Carrillo et al. propose the use of deuterated phthalates as internal standards for the accurate determination of phthalates in wine by headspace solid-phase micro-extraction followed by GC-MS.113 Unlike other internal standards proposed previously, such as benzyl benzoate, deuterated phthalates enabled matrix error-free determinations to be performed without standard-addition, because statistically equal slopes were obtained for synthetic, white, rosé and red wines. Quantification in complex samples using SPME-based methods is usually difficult, because matrix components affect sensitivity and due to the lack of certified reference materials for calibration of the method. The standard-addition method is the most commonly used calibration method when quantification is prone to matrix-effect errors. However, this calibration method is time-consuming, because a calibration graph is necessary for each sample. The internal standard method is an alternative option, although the need to find a suitable internal standard for the target compounds is an important disadvantage. This compound should be as similar as possible to the analyte measured, as sources of variation should affect both analyte and internal standard in the same way, and should not be present in the matrix. Accuracy and precision are considerably improved when isotopically labeled standards are used in mass spectrometry determinations, because these compounds have similar physical and chemical properties to their unlabeled analogues. This method is shown to be free of matrix-effect errors and therefore calibration can be performed using standard solutions in synthetic wine. Therefore, it has advantages over other methods, because the time and work required for analysis are significantly reduced as calibration by standard-addition is avoided. Holadová et al.114 developed a HS-SPME method comparing the efficiency of three different fibers (silica fibers coated with polydimethylsiloxane (PDMS), polyacrylate (PA), Carboxen/polydimethylsiloxane (CX/PDMS), and polydimethylsiloxane/divinyl benzene (PDMS/DVB)). This developed SPME method, employing a PDMS 100 fiber together with methanol as a matrix modifier, enables headspace phthalate determination in vegetable oil samples. However, reliable results can only be obtained by maintaining careful temperature control and employing intensive stirring with a magnetic stirrer. Rios et al.115 also tested three different fibers (polyacrylate (PA), divinyl benzene–Carboxen–polydimethylsiloxane (DVB/CAR/PDMS), and polydimethylsiloxane (PDMS)). Their main findings are: PA fibers are not suitable for the lightest polar phthalates, which exhibited poor extraction and repeatability values; PDMS fibers give a very poor response for some of the heavier non-polar phthalates; whereas DVB/CAR/PDMS fibers exhibit the best response and repeatability values for the majority of the phthalates studied. The main benefit of the analytical method proposed is the absence of sample manipulation and hence the avoidance of possible contamination arising from glassware, the environment, solvents and samples. PTFE/silicon was used by Al-Saleh et al.116 for determining five PAEs in waters in plastic bottles available in Saudi markets, whereas Zhao et al.117 investigated a HS-SPME method based on a polyaniline–polypyrrole (PANI–PPY) composite film coated on a stainless steel wire and prepared by cyclic voltammetry for determining PAEs in beer, and Yang et al.118 determined PAEs in dried spikes of Prunella vulgaris.
| Phthalate | Sorbent/time | Matrix | r2 | Repeatability | LOD/LOQ | Recovery | Ref. |
|---|---|---|---|---|---|---|---|
| DBP, DEP, DEHP | 60 min | Cow milk | — | 3.9–12 | 5–45a/310–330b | 82–104 | 110 |
| DEP, DBP, DEHP | PTFE/silicone/60 min | Human milk | — | — | 120b–1200/— | — | 111 |
| DMP, DEP, DBP, BBP, DEHP, DOP | CW–DVB and PDMS–DVB/10 min | Red, white and rosé wines | 0.922–0.999 | 2–28 | 60–2900/100–4200c | 88–124 | 112 |
| DMP, DEP, DBP, DEHP, BBP, DOP | PDMS–DVB/10 min | Synthetic, white, rosé and red wines | 0.995–0.999 | 0.24–4.6 | 5–16/21–400c | — | 113 |
| DMP, DEP, DBP, DAP, DNOP | PDMS–DVB/10 min | Vegetable oil | 0.940–0.999 | 14–23 | 0.06–0.3/0.2–0.5b | — | 114 |
| DMP, DEP, DiBP, DBP, BBP, DEHP, DNP, DOP | DVB/CAR/PDM/30 min | Virgin olive oils | 0.975–0.999 | 7.7–18.7 | 0.02–0.05/—b | — | 115 |
| DMP, DEP, DBP, BBP, DEHP | PTFE + silicon/10 min | Bottled water | 0.9981–0.999 | 3.2–12.6 | 502–856/825–1321c | 95.8–113.8 | 116 |
| DMP, DEP | PANI–PPY/10–50 min | Beer | 0.9929–0.9985 | 3.6–6.1 | 50–380/—c | 97–100 | 117 |
Liquid–liquid extraction method
Liquid–liquid extraction (LLE), also known as solvent extraction and partitioning, is a method for separating compounds based on their relative solubility in two different immiscible liquids. The advantages of this method, compared to the drawbacks introduced, include its simplicity, precision, low LODs, high recovery and cost-effectiveness. In addition, common organic solvents are used in traditional LLE without further cleanup. This method is convenient and provides accurate and reliable results. This well-known method has been applied to PAE determination in different matrices using, essentially, acetonitrile or hexane as extraction solvent, whereas few papers employ ethyl acetate, dichloromethane or methylene chloride–petroleum ether. Table 5 summarizes all the analytical parameters reported.| Phthalate | Method | Matrix | r2 | Repeatability | LOD/LOQ | Recovery | Ref. |
|---|---|---|---|---|---|---|---|
| 12 PAEs | Acetonitrile + SPE (florisil) | Oily foods | >0.998 | <20 | —/0.050–0.1 | 72–105 | 119 |
| 15 PAEs | Acetonitrile | Olive oils | 0.9922–0.9984 | 2.52–10.28 | 0.003–1.2/0.010–4.0 | 93.5–101.3 | 120 |
| DBP | Hexane | Indian tobacco | — | — | — | — | 121 |
| DBP, DiBP, DINP, DEHP | Hexane | Vegetable oils | 0.9996a | <25 | 0.01–1/0.04–3 | 82–106 | 122 |
| DMP, DEP, DIBP, DnBP, BBP, DCHP, DEHP, DNP, DnOP | Hexane | Drinking water, food | >0.98 | 3.8–20.5 | — | 54.7–104.2 | 123 |
| 22 PAEs | Hexane | Honey, royal jelly | 0.9990–0.9999 | <11.8 | 0.3–1.2/1.0–10.0c,e | 80.1–110.9 | 124 |
| 1.5–15/5.0–50.0d,e | |||||||
| DMP, DEP, DBP, BBP, DEHP, DOP | Methylene chloride–petroleum ether (20 + 80, v/v) | Jordanian bottled water | 0.9803–0.9983 | 1.7–3.9 | 20–80b/— | 84.6–96.4 | 126 |
Regarding acetonitrile as extraction solvent, two papers were published between 2010 and 2013. In the first paper119 the author develops a method for analyzing 12 PAEs along with four other plasticizers (dibutyl sebacate (DBS), acetyl tributyl citrate (ATBC), di-(2-ethylhexyl) adipate (DEHA), di-isononyl cyclohexane-1,2-dicarboxylate (DINCH)) in vegetable oil, pesto, and tomato sauce. The method consists of two steps: i) extraction performed using acetonitrile (60 mL per 10 g sample) with NaCl addition, centrifugation, evaporation to dryness under a stream of nitrogen and dissolving the residue with 2 mL hexane; and ii) cleanup using a SPE column (LC-Florisil glass containing Florisil), where plasticizers are eluted with acetone-n-hexane (10 + 90, v/v), the collected eluate is evaporated to dryness under a stream of nitrogen and the residue is dissolved in acetonitrile. In the paper, the author shows how plasticizers were recovered from oily foods by simple L–L extraction with acetonitrile but, at the same time, because of the different polar properties of the 12 PAEs and other plasticizers, some of them were not extracted quantitatively by this procedure (DEHP, DEHA, DOP, DINP, and DIDP). So, a SPE method (Florisil as SPE stationary phase) was also used as an additional cleanup for eliminating interference by co-extracted substances. After this step, the phthalates and the other four plasticizers were detected in the same GC run. The main results exhibit good linearity up to 2.5 μg g−1 in most cases, except for DINP, DINCH, and DIDP (200 μg g−1), with correlation coefficients >0.998 in all cases, whereas recoveries are in the range 72–105% and the repeatability as RSD <20% and LOQ values ranging between 0.050 and 0.1 μg g−1, except for DINP and DIDP (2 μg g−1).
In the paper by Dugo et al.,120 the extraction was performed using acetonitrile (1 mL per 2 g sample) and, after centrifugation at 5000 rpm for 10 min, the collected phase was analyzed by high-resolution gas chromatography coupled with mass spectrometry (HRGC-MS), providing good analytical parameters for analyzing PAEs in olive oils from Sicily and Molise (Italy). Particularly interesting is the chemometric approach reported (i.e., a Mann–Whitney non-parametric test of differences between groups and a factor analysis with principal components): starting from the PAE data, the authors managed to identify different types of production area of olive oils or different years of production.
More extensive is the literature regarding the use of hexane as extraction solvent: 2 papers in 2011 and 3 papers in 2014. Among 21 components of Lobelia inflata (Indian tobacco), Joshi et al.121 determined DBP, but no information on analytical parameters is present. The paper by Nanni et al.122 clarifies certain aspects of vegetable oil contamination by PAEs. After extraction with hexane, the authors found low levels of DEHP, DINP, DBP and DIBP in vegetable oils. In Italy, olive-derived oils are the oils most consumed, and clarification of the reasons for such contamination could be relevant in order to reduce PAEs levels and to improve the quality of national oil production. Das et al.123 set up a suitable approach for developing an approximate estimation of the exposure of the South Delhi population to fifteen different PAEs. They investigated the PAE content in indoor air, outdoor air, indoor dust, outdoor dust, drinking water and food; for food matrices, they employed L–L extraction with hexane whereas the other matrices were processed by SPE with XAD-2. An interesting application is presented by Zhou et al.,124 where a protocol to analyze 22 PAEs in honey and royal jelly is reported. The method provides a wide linear range, satisfactory precision, accuracy, no significant matrix effect, and acceptable recovery and the results (seven PAEs in honey samples and five PAEs in royal jelly samples) suggest that PAEs could migrate from the surroundings into honey and royal jelly samples. In addition, extraction with hexane followed by GC-MS analysis was also used by Xu et al.125 for comparing the new method (developed by them) for determining DBP in white wine based on an indirect competitive enzyme linked immunosorbent assay (icELISA).
Finally, a paper from Zaater et al.126 presents PAE determination in Jordanian bottled water performed by LLE using methylene chloride–petroleum ether (20 + 80, v/v) and comparing, inter alia, LL-GC-MS and LL-HPLC-UV procedures.
Dispersive liquid–liquid (micro) extraction method
Considering the complexity of sample matrices and the relatively low concentration of PAEs, sample pretreatment and enrichment processes are crucial steps in the analytical procedure to obtain accurate and sensitive results. In the previous paragraphs, we reported traditional pretreatment methods, such as LLE and SPE, which are time-consuming and need a large amount of organic solvents, which are dangerous for human health and the environment. Research has been oriented towards the development of simple, economical, and miniaturized pretreatment methods. Recently, a novel micro-extraction technique termed dispersive liquid–liquid micro-extraction (DLLME) was developed by Assadi et al.127 This is based on a ternary solvent system, as used in homogeneous liquid–liquid extraction and cloud point extraction.128,129 In this method, the appropriate mixture of extraction solvent and dispersive solvent is injected rapidly into an aqueous sample by a syringe to form a cloudy solution, which markedly increases the contact surface between phases and reduces extraction times, with an increase in enrichment factors. The method is based on the addition of an immiscible solvent with higher density (chlorinated solvents) to aqueous samples for the extraction step. The dispersant solvent plays an important role: it increases contact between two immiscible solvents, forming an interface which is crucial in the extraction process. After extraction, phase separation is performed by centrifugation and analytes in the sediment phase are determined by chromatography or spectrometry methods.130–132 The advantages of DLLME are its simplicity, rapidity, low cost, high recovery and enrichment factors.The first application of a DLLE method to PAEs is dated 2010 and regards the determination of such compounds (particularly DBP, BBP, DIOP, and DNOP) in a bottled water sample by means of a modified DLLE technique, i.e. ultrasound-assisted dispersive liquid–liquid micro-extraction (UA-DLLME), followed by GC-flame ionization detection (GC-FID).133 The authors investigated various parameters affecting the whole analytical procedure: the effect of the extraction solvent (i.e., dichloromethane CH2Cl2, 1,2-dichloroethane C2H4Cl2, 1,1,2,2-tetrachloroethane C2H2Cl4, chlorobenzene C6H5Cl, tetrachloroethylene C2Cl4, and carbon tetrachloride CCl4) with their relative volumes (from 10 to 70 μL), the effect of different dispersant solvents (i.e., methanol, ethanol, acetonitrile, THF, acetone and isopropanol) with their relative volumes (i.e., 0.2, 0.4, 0.5, 0.6, 0.8 and 1.2 mL), and the effects of ultrasonication time, ionic strength and pH. Under optimum conditions (20 μL CCl4 as extraction solvent, 0.8 mL methanol as dispersant solvent, 2.0 min ultrasonication time, 5.0% sodium chloride, pH not important, between 3.0 and 9.0), enrichment factors of phthalates ranged from 490- to 530-fold and recoveries ranged from 84.8% to 104.7%. A good linear relationship between the peak area and the concentration of analytes was obtained in the range 6.9–444 mg L−1 (r2 > 0.9992); LODs and LOQs varied between 1.0 and 1.2 μg L−1 and between 3.4 and 4.5 μg L−1, respectively. Intra-assay and inter-assay precision, expressed as the relative standard deviation, were in the ranges 1.4–2.0% and 3.0–3.7%, respectively.
Until 2013, only one paper was published regarding a modified DLLE method followed by GC-MS analysis for food matrices. On the other hand, between 2013 and 2014, in just two years, seven papers were published regarding determination of PAEs by a modified DLLE method followed by GC-MS. In particular, two of them are devoted to PAE determination in bottled and drinking waters,134,135 four in soft drinks, wine and high-alcohol beverages72,136–138 and one in river water samples.139 Table 6 summarizes the main parameters reported in these papers, such as the correlation coefficients, reproducibility, enrichment factors, LODs, LOQs and recoveries.
| Phthalate | Method | Matrix | r2 | Repeatability | EF | LOD/LOQ | Recovery | Ref. |
|---|---|---|---|---|---|---|---|---|
| DMP, DEP, DBP, BBP, DEHP, DOP | LDS-VSLLME-GC-MS | Bottled water | 0.9823–0.9992 | 0.80–11.9 | 200–290 | 8–25/— | 77.4–106.6 | 134 |
| DMP, DEP, DBP, BBP, DEHP, DOP | DLLME-GC-MS | Bottled water | 0.995–0.999 | 1.31–5.22 | 220b | 5–22/— | — | 135 |
| DMP, DEP, DBP, BcEP, BBP, DEHP | USVADLLME-GC-MS | Alcoholic (10–40% alcohol by volume) | 0.9446–0.9938 | 4.9–7.0 | 220–300 | 22–100/75–335 | 91.5–100.5 | 72 |
| DMP, DEP, DiBP, DBP, BcEP, BBP, DEHP | USVADLLME-GC-MS | Alcoholic (<6% alcohol by volume), soft drinks | 0.9235––0.9959 | 5.9–7.6 | 205–315c | 30–100/110–280 | 94.2–99.6c | 136 |
| 172–285d | 95.6–99.4d | |||||||
| DBP, BBP, DEHP, DNOP | VSLLME-GC-MS | Chinese liquor | 0.9938–0.9971 | 6.8–11.2 | 140–184 | 4.9–13/— | 81.2–93.7 | 137 |
| DMP, DEP, DiBP, DBP, DEHA, DEHP | SB-DLLME-GC-MSa | Mineral water, lemon juice, cola, vinegar | 0.996–0.999 | 3.0–5.2 | 266–556 | 90–250/310–850 | 49–100 | 138 |
| DMP, DEP, DBP, BBP, DEHP, DOP | VA-μ-SPE-LDS-DLLME-GC-MS | River water | 0.9906–0.9952 | 4.9–8.0 | — | 6–20/30–70 | 89.7–104.1 | 139 |
| DMP, DEP, DiBP, DBP, DEHP | HLLE-GC-FID | Mineral water, cola, vinegar | 0.994–0.999 | 3–8 | 172–309 | 0.02–0.71/0.05–2 | 81.3–110.0 | 143 |
| DMP, DEP, DBP, BBP, DEHP, DOP | Automated DLLME-GC-MS | River water and tap water | 0.9903–0.9978 | 3.5–5.9 | 178–272 | 10–20/— | 88.5–112.8e | 144 |
| 84.1107.6f | ||||||||
| DMP, DAP, DBP, BBP, DCHP, DEHP | DLLME-GC-MS | Drinking water | 0.9901–0.9962 | 4.6–6.8 | 681–889 | 2–8/— | 68.1–88.9 | 145 |
| DMP, DEP, DAP, DBP, BBP, DCHP, DEHP | LPME-GC-MS | Mineral water, tap water | 0.9940–0.9971 | 5.5–7.7 | 307–412 | 20–50 | 84–115 | 146 |
Mousa et al.135 applied a dispersive liquid–liquid micro-extraction method followed by GC-MS analysis (DLLME-GC-MS) for PAE determination in bottled drinking water samples. Under optimized conditions (500 μL xylene as extraction solvent, 20 μL methanol as dispersive solvent, sonication time 20 min, pH 2), very good linearity is observed for all analytes in a range between 0.05 and 150 μg L−1, with correlation coefficients (r2) between 0.995 and 0.999. The LODs are 0.005–0.22 μg L−1. The authors evaluate the DLLME reproducibility, as RSD, to be 1.3–5.2%. Actually, the authors deal with a very interesting issue: understanding the leaching profile of phthalates from bottled water when the bottles are exposed to direct sunlight during summer (temperature 34–57 °C) and sampled at different intervals. Results show that di-n-butyl, butyl benzyl, and di-2-ethylhexyl phthalate leach from bottles for up to 36 h. Thereafter, degradation of phthalates is also observed.
Two interesting modified DLLE methods are proposed by Zhang and Lee134 and Russo et al.72,136 Zhang and Lee developed novel low-density solvent-based vortex-assisted surfactant-enhanced-emulsification liquid–liquid micro-extraction (LDS-VSLLME) for the fast, simple and efficient determination of six phthalate esters (DMP, DEP, DnBP, BzBP, DEHP, and DnOP) in bottled water samples followed by GC-MS. In the extraction procedure, an aqueous sample solution is injected into a mixture of extraction solvent (toluene) and surfactant (cetyltrimethylammonium bromide) to form an emulsion with the assistance of vortex agitation. After extraction, phase separation by centrifugation, and removal of the spent sample, the toluene extract is collected and analyzed by GC-MS. The addition of a surfactant enhances dispersion of the extraction solvent in the aqueous sample. Using a relatively less toxic surfactant as an emulsifier overcomes the disadvantages of traditional organic dispersive solvents that are usually highly toxic and expensive. With the aid of a surfactant and vortex agitation to achieve good dispersion of the organic extraction solvent, extraction equilibrium is achieved within 1 min. Another prominent feature of the method is the simple procedure for collecting a solvent less dense than water by a micro-syringe. After extraction and phase separation, the aqueous sample is removed using a 5 mL syringe. As reported by the authors, this method simplifies the use of low-density solvents in DLLME. Under optimized conditions, the proposed method provides good linearity in the range 0.05–25 μg L−1, low limits of detection (0.008–0.025 μg L−1) and good enrichment factors up to 290.
Russo et al. apply an ultrasound-vortex-assisted dispersive liquid–liquid micro-extraction (USVADLLME) procedure coupled with gas chromatography-ion trap mass spectrometry (GC-IT/MS). Actually, ultrasound-assisted (USADLLME) and vortex-assisted emulsification (VADLLME)-based protocols have recently been proposed.140–142 These approaches reduce the volume of organic solvent required and simultaneously improve extraction efficiency without the use of a dispersive solvent. In fact, in the two papers the authors investigate the use of a dispersive solvent in alcoholic matrices (beer, wine, and liquors where the alcohol content ranges between 4 and 40% alcohol by volume) and soft drinks (soda, cola, tonic, sprite, 7 up). In the presence of alcoholic beverages, no dispersive solvent is added because the investigated matrix has an alcohol content that exerts a co-surfactant effect, and because the ultrasound used provides sufficient energy to obtain the finely dispersed phase required to quantitatively extract the solutes. On the other hand, in a soft drink a dispersive solvent is necessary because of the absence of alcohol: ethanol (4%, v/v) is used as the dispersive solvent in these cases. Use of a vortex is essential because it initially helps to disperse the extracting solvent. When a vortex is not used, the extracting solvent forms a biphasic system, where the phase with higher density is an emulsion. The authors investigate and optimize different parameters, like the extraction solvents, the number of repeated extraction steps and the isolation technique. The extraction efficiency is optimized by varying the extractants dichloromethane, chloroform, tetrachloroethylene and carbon tetrachloride. Homogenization, shaking, or ultrasonication is regarded as the second factor in optimizing the isolation of phthalates from matrices. In addition, microwave-assisted extraction is performed before GC-MS determination. In this way, very good LODs (≥0.022 μg L−1) and LOQs (≥0.075 μg L−1) are reached with recoveries ranging from 85% to 100.5% and enrichment factors from 220-to 300-fold.
Two papers use a vortex-assisted DLLME method for analyzing PAEs in river water samples139 and Chinese liquor samples.137 In particular, in the latter paper, the authors employ a high-density solvent (carbon tetrachloride) as extraction solvent for the pretreatment of four PAEs in Chinese liquor samples prior to quantification by GC-MS. The high-density extraction solvent helps produce better dispersion with the aid of a surfactant and vortex agitation, resulting in rapid extraction equilibrium within only 30 s. The authors investigate and optimize various experimental factors, such as the type and volume of the extraction solvent, the type and concentration of the surfactant, the extraction time, sample pH, and addition of salt.
Even if the paper by Guo and Lee139 does not concern a food matrix (river water samples), it is interesting because of the entire procedure involved (vortex-assisted micro-solid-phase extraction, (VA-μ-SPE), followed by low-density solvent-based dispersive liquid–liquid micro-extraction (LDS-DLLME), followed by GC-MS) and because of the particularly good LODs attained (0.006–0.02 μg L−1) and LOQs (0.03–0.07 μg L−1). The analytes are first extracted and pre-concentrated by vortex-assisted μ-SPE, which is faster than conventional μ-SPE (6 min compared to 30 min), and then desorbed by ultrasonication into acetonitrile. The latter serves as the dispersive solvent in the subsequent LDS-DLLME step that further pre-concentrates the analytes. The authors analyze some key parameters for VA-μ-SPE and LDS-DLLME, such as the selection and amount of sorbent, vortex time, ultrasonication solvent and time, type and volume of extraction solvent for DLLME, and the speed and time of centrifugation.
Farajzadeh et al.138 developed a green analytical method for analyzing PAEs in mineral water, lemon juice, beverage (cola), and vinegar packed in plastic containers. In particular, they propose a solid-based disperser liquid–liquid micro-extraction (SB-DLLME) method, in which the dispersive solvent is replaced by a sugar cube as a solid disperser and the consumption of toxic organic solvents is minimized. By manual shaking, the sugar is dissolved and the extractant is released into the aqueous phase as very small droplets to provide a cloudy solution. Under optimized conditions, the proposed method achieved good precision (RSD less than 5.2%), high enrichment factors (266–556), and low LODs (0.09–0.25 μg L−1). The method was successfully applied for PAEs determination in different samples, and good recoveries (71–103%) were achieved for spiked samples.
In 2013, Farajzadeh et al. published another paper concerning L–L extraction using acetonitrile in mineral water, cola and vinegar by a simple and rapid method.143 In fact, the authors employ homogeneous (water and acetonitrile) liquid–liquid extraction based on phase separation in the presence of a salt (NaCl) performed in a narrow-bore tube (HLLE). Actually, the analysis takes place by GC-flame ionization detection (FID), but we believe this to be an interesting method so we would like to review it, considering the advantages, such as a relatively short analysis time and high EFs, offered by this technique.
Particular attention should be addressed to another paper by Guo and Lee.144 They developed an innovative automated procedure, low-density solvent-based/solvent demulsification dispersive liquid–liquid micro-extraction (automated DLLME) coupled to GC-MS analysis. The most significant innovation of this method is the automation of the entire procedure, including the extraction of model analytes (phthalate esters) by DLLME from an aqueous sample solution, breaking-up of the emulsion after extraction, collection of the extract, and analysis of the extract by GC-MS. No human intervention is required in the entire extraction procedure, except at its beginning. The application of a low-density solvent as extraction solvent and a solvent demulsification technique to break up the emulsion simplifies the procedure and facilitates its automation. Enrichment factors of between 178- and 272-fold were obtained for phthalate esters along with good LODs (ranging between 0.01 and 0.02 μg L−1) and satisfactory repeatability (RSD < 5.9%).
Finally, two interesting papers describing a screening method for analyzing PAEs in water are presented by Farahani et al.145,146 The authors use a liquid-phase micro-extraction (LPME) technique using a directly suspended organic micro-drop coupled with capillary GC-MS. Actually, LPME is a fairly new method of sample preparation.147 It is a miniaturized adaptation of conventional LLE, where only microliters of the solvents are used. This quantitative LPME method is a green and satisfactory analytical procedure, for which excellent accuracy and precision have been demonstrated, as it is simpler and more convenient, compared with conventional sample preparation methods. The approach proposed by Farahani et al. offers different advantages such as simplicity, low cost, ease of operation, no possibility of sample carry-over and high enrichment factors. In addition, the technique only requires a small volume (7 μL) of organic extractant (1-dodecanol), being therefore an environmentally friendly approach. The authors propose that this method could be routinely used for screening purposes.
Other sample extraction/preparation methods
Sonication/ultrasonication extraction method
This method is based on the application of sound energy to agitate particles in a sample. Ultrasonic frequencies (>20 kHz) are usually used, leading to an ultrasonication process. Actually, this process is not much present in the literature when applied to food matrices. In fact, interesting applications are found regarding plastic articles,148 dust,149 polymer materials150–153 and sludge from wastewater treatment plants,154,155 whereas only one paper based on sample extraction/preparation by sonication/ultrasonication156 followed by GC-MS analysis is present in the literature. It should be considered that most of the time sonication/ultrasonication is coupled to LLE,157,158 SPE/SMPE139 and DLLE72,136 extraction procedures for attaining high sensitivity and recoveries, but these methods have just been reviewed above.Shen's paper156 regards determination of PAE in plastic products for food use, including packaging bags, packaging film, containers, boxes for microwave oven use, straws, spoons, cups, plates, etc. In a glass bottle, 1 g sample is soaked in 10 mL hexane for 30 min, followed by sonication for 10 min: this process helps improve the contact between solvent and sample. The author also tried different solvents, i.e., hexane, acetone and water. Acetone as extractant provides good recoveries (average recoveries 126.2% with RSD 12.5), but many interfering compounds are co-isolated, which does not allow PAEs to be determined at low levels; on the other hand, water does not provide good recoveries (average recoveries 78.5% with RSD 9.8). Hexane provides good recoveries (average recoveries 90.5% with RSD 8.5) and allows phthalates to be detected at a level of 10.0 μg kg−1. This procedure is applied twice and afterwards the two fractions are combined. The extract is then reduced to dryness (with liquid nitrogen) and, after a cleanup procedure (performed by SPE with a LC-C18 cartridge), analyzed by GC-MS. The method has successfully been applied to 25 real samples: only one was found to be free of phthalates, whereas the other 24 samples were found to contain at least three phthalates (predominantly DEHP).
Thermal desorption extraction method
Thermal desorption is an environmental remediation technology that utilizes heat to increase the volatility of contaminants so that they can be removed from a matrix: the volatilized contaminants are then either collected or thermally destroyed. A thermal desorption system therefore has two major components: the desorber itself and an off-gas treatment system. This method is employed to investigate semi-volatile organic compounds with boiling points ranging between 240 and 400 °C,159 such as phthalate esters, and in meaningful quantities both in the gas phase and on the surface of the solid phase (e.g., particles). For these reasons applications mainly concern air matrices,160–164 whereas no applications are reported in the literature for a food matrix. Actually, a few papers (5) show how thermal desorption-gas chromatography-mass spectrometry (TD-GC-MS) could be used for analyzing extracts of real water samples (i.e., industrial ultrapure waters, sea and estuarine waters, lake water, river water and wastewater) produced by stir bar sorptive extraction,165–167 membrane-assisted solvent extraction167 or solid (micro)phase extraction.168,169 Even if the samples are not drinking water, we believe it to be important to report the main findings in these papers as evidence for this technique. Most of the time the authors use TD-GC-MS for analyzing PAEs among different persistent organic pollutants (POPs) (i.e., polycyclic aromatic hydrocarbons, polychlorinated biphenyls, nonyl phenols, polybrominated biphenyls, polybrominated diphenyl ethers, and insect repellents such as permethrin, N,N-diethyl-m-toluamide, and icaridin) present in the samples. The TD-GC-MS method, based on the different boiling points of each compound, is very useful for this purpose. Coupling between different extraction methods (i.e., SPE, SPME, SBSE, MASE) and TD-GC-MS allows very low LODs and LOQs to be attained (<0.25 μg L−1 and < 0.5 μg L−1, respectively), whereas recoveries are not always so good (for instance, DMP166 recovery 29%, or DMP and DEP,168 both polar phthalates, 0% and 32%, respectively) (Table 7). Prieto et al.167 compared two different extraction methods (SBSE and MASE): the SBSE method provided better LOD values, whereas MASE resulted in similar recoveries, but faster extraction. The authors give evidence that polypropylene membranes (in MASE) have the advantages of low cost and easy handling, although a thorough cleaning procedure is necessary in order to improve the LODs in PAE determination: this is due to the composition of the membrane material. The results obtained by both methodologies are comparable, indicating that these efficient and environmentally friendly analytical procedures can be employed for determining priority organic contaminants in water samples. On the other hand, the paper by Liu et al.169 regards a procedure modified from the conventional SPE method. The authors propose a method for sampling water through a sampling tube containing a hydrophobic sorbent (Tenax TA) to concentrate aqueous phthalate esters. This paper is also interesting because of the equipment used by the authors: in fact, a solid trap is demoisturized by two-stage gas drying (Automated Thermal Desorption, ATD) before being subjected to GC-MS. In the first stage, a sample is thermally extracted from a solid extraction tube and carried by a reverse gas flow (helium) into a cold trap to be re-concentrated; in the second stage, the concentrated specimens are thermally desorbed again and transferred by helium gas into a capillary column as a narrow band to improve the sensitivity of the chromatography. The process allows avoidance of the solvent extraction procedure necessary for the conventional SPE method, and, primarily, permits automation of the analytical procedure for high-volume analyses. The authors investigated several parameters (i.e., desorption time and temperature, the type and dosage of the sorbent material), obtaining LODs for five phthalates (DEP, DBP, BBP, DEHP, and DOP) between 36 ng L−1 and 95 ng L−1 and recovery rates between 15% and 101%.| Phthalate | Method | Matrix | r2 | Repeatability | LOD/LOQ | Recovery | Ref. |
|---|---|---|---|---|---|---|---|
| DMP, DEP, DBP, BBP, DEHP, DOP | SBSE-TD-GC-MS | Drinking water | 0.80–0.99 | 4–20 | 0.1–6.3/0.3–19 | 95–124 | 165 |
| DMP | SBSE-TD-GC-MS | Water | 0.999 | 11 | 150/328 | 29 | 166 |
| DMP, DEP, DBP, BBP, DEHP, DOP | SBSE-TD-GC-MS | Water | >0.99 | 3–19 | 0.1–10.0/0.6–41.9 | 91–126 | 167 |
| DMP, DEP, DBP, BBP, DEHP, DOP | MASE-LVI-PTV-GC-MS | Water | >0.99 | 3–19 | 29.8–252/63.6–475 | 81–117 | 167 |
| DMP, DEP, DBP, BBP, DEHP, DOP | SPME-TD-GC-MS | Water | — | 2–10 | —/50–200 | 0–116 | 168 |
| DEP, DBP, BBP, DEHP, DOP | SPE-ATD-GC-MS | Ultrapure water | 0.9915–0.9956 | 5.9–10.1a | 36–95/— | 15–101 | 169 |
Sample extraction and preparation using Soxhlet
This old extraction method is used when the desired compound has limited solubility in a solvent, and the impurity is insoluble in that solvent. Its advantage is that it allows unmonitored and unmanaged operations, while efficiently recycling a small amount of solvent to dissolve a larger amount of material. Nowadays, the Soxhlet procedure is a little-used analytical process for analyzing food matrices. In fact, according to the Scopus database only 40 papers relate to PAE determination using Soxhlet and GC-MS analysis, but all of these are aimed at investigating PAEs in plastics/precious articles,148,170 indoor air dust,171–173 medicinal leaf extracts,174–178 textile materials,179 soil180,181 and sewage sludge.182 All the papers perform the Soxhlet procedure using cyclohexane, dichloromethane or methanol, with an extraction time ranging between 4 h and 24 h at room temperature, or 12 h at 60 °C. The LODs are quite good, around 1 μg g−1, but in some cases the authors also manage to reach very low levels, i.e. 0.009 μg g−1.173 Actually, the only application related to food matrices is dated 1998: Spanish authors183 determined dimethyl phthalate (DMP) and diethyl phthalate (DEP), along with other organic compounds, in drinking water. The extraction occurred by Soxhlet using dichloromethane for 48 h, whereas the analysis was performed by high-performance gas chromatography coupled with low-resolution mass spectrometry (HPGC-LRMS). No information on the analytical parameters is reported in the text, so it is quite difficult to evaluate the procedure.Finally, even though no food matrix is involved, it is worth mentioning the paper by Nerin et al.,184 where optimization of supercritical fluid extraction (SFE) using CO2 for PAE extraction from 15 samples of recycled paper and board (P & B) has been studied. As reported by the authors, SFE is a good procedure for extracting PAEs, being easy to use with minimum handling and time; a lower amount of organic solvents (<2 mL) is required, confirming SFE as a safer and environmentally friendly technique. The collection system for the final extract from SFE is critical, and a polar solvent such as methanol is required to elute the more polar compounds. In this way, SFE extraction provides results similar to those obtained by liquid extraction using ethanol.
Conclusions
All the analytical methods reported in this review are useful for determining PAEs in food matrices, both in terms of reproducibility and accuracy. The big issue regarding contamination is relevant, but in the literature there are at present different methods to solve it. In our opinion, we believe that future developments could envisage the possibility of miniaturizing all the procedure or totally automating the extraction step and the GC-MS analysis as well.References
- D.-P. Xu, S. Li, Y.-H. Chen, H.-B. Li, A.-N. Li and X.-R. Xu, Int. J. Mod. Biol. Med., 2013, 4, 12 Search PubMed
.
- O. Lau and S. Wong, J. Chromatogr. A, 1996, 737, 338 CrossRef CAS
- M. C. Yin and K. H. Su, J. Food Drug Anal., 1996, 4, 313 CAS
- D. Balafas, K. J. Shaw and F. B. Whitfield, Food Chem., 1999, 65, 279 CrossRef CAS
- K. Inoue, M. Kawaguchi, F. Okada, Y. Yoshimura and H. Nakazawa, Anal. Bioanal. Chem., 2003, 375, 527 CAS
- B. Tienpont, F. David, E. Dewulf and P. Sandra, Chromatographia, 2005, 61, 365 CAS
- ATSDR, Toxicological profile for diethylphthalate, Agency for Toxic Substances and Disease, Atlanta, GA, 1995 Search PubMed
- ATSDR, Toxicological profile for di-n-octylphthalate, Agency for Toxic Substances and Disease Registry, Atlanta, GA, 1997 Search PubMed
- ATSDR, Toxicological profile for di-n-butyl phthalate, Agency for Toxic Substances and Disease, Atlanta, GA, 2001 Search PubMed
- ATSDR, Toxicological profile for di(2-ethylhexyl) phthalate, Agency for Toxic Substances and Disease, Atlanta, GA, 2002 Search PubMed
- NTP-CERHR, NTP-CERHR expert panel report on di(2-ethylhexyl) phthalate, NTP-CERHR-DEHP, 2000 Search PubMed
- NTP-CERHR, Monograph on the potential human reproductive and developmental effects of di-isononyl phthalate (DINP), NIH Publ. No. 03–4484, 2003 Search PubMed
- NTP-CERHR, Monograph on the potential human reproductive and developmental effects of di-isodecyl phthalate (DIDP), NIH Publ. No. 03–4485, 2003 Search PubMed
- NTP-CERHR, Monograph on the potential human reproductive and developmental effects of di-n-butyl phthalate (DBP), NIH Publ. No. 03–4486, 2003 Search PubMed
- NTP-CERHR. Monograph on the potential human reproductive and developmental effects butyl benzyl phthalate (BBP). NIH Publ. No. 03–4487, 2003 Search PubMed
- NTP-CERHR, Monograph on the potential human reproductive and developmental effects of di-n-octyl phthalate (DnOP), NIH Publ. No. 03–4488, 2003 Search PubMed
- NTP-CERHR. Monograph on the potential human reproductive and developmental effects of di-n-hexyl phthalate (DnHP). NIH Publ. No. 03–4489, 2003 Search PubMed
- NTP-CERHR, Expert panel re-evaluation of DEHP, Meeting summary, October 14, 2005, http://www.noharm.org/details.cfm?ID=1130%26;type=document Search PubMed
- U. Heudorf, V. Mersch-Sundermann and J. Angerer, Int. J. Hyg. Environ. Health, 2007, 210, 623 CrossRef CAS PubMed
- P. Vera, M. Aznar, P. Mercea and C. Nerín, J. Mater. Chem., 2011, 21, 420 RSC
- M. Aznar, P. Vera, E. Canellas, C. Nerín, P. Mercea and A. Störmer, J. Mater. Chem., 2011, 21, 4358 RSC
- I. Rusyn and J. C. Corton, Mutat. Res., Rev. Mutat. Res., 2012, 750, 141 CrossRef CAS PubMed
- B. Gevao, A. N. Al-Ghadban, M. Bahloul, S. Uddin and J. Zafar, Indoor Air, 2013, 23, 126 CrossRef CAS PubMed
- T. J. U. Wams, Sci. Total Environ., 1987, 66, 1 CrossRef CAS
- WHO, Environmental Health Criteria 131, 1992 Search PubMed
- C. G. Bornehag, J. Sundell, C. J. Weschler, T. Sigsgaard, B. Lundgren, M. Hasselgren and L. Hägerhed-Engman, Environ. Health Perspect., 2004, 112, 1393 CrossRef CAS
- J. H. Petersen and L. K. Jensen, Food Addit. Contam., Part A, 2010, 27, 1608 CrossRef CAS PubMed
- W. W. Hubert, B. Grasl-Kraupp and R. Schulte-Hermann, Crit. Rev. Toxicol., 1996, 26, 365 CrossRef PubMed
- C. Simoneau and P. Hannaert, Food Addit. Contam., 1999, 16, 197 CrossRef CAS PubMed
- M. Vitali, M. Guiditti, G. Macilenti and C. Cremisini, Environ. Int., 1997, 23, 337 CrossRef CAS
- M. A. Kamrin, J. Toxicol. Environ. Health, Part B, 2011, 12, 157 Search PubMed
- A. P. van Wezel, P. van Vlaardingen, R. Posthumus, G. H. Grommentijn and D. T. H. Sijm, Ecotoxicol. Environ. Saf., 2000, 46, 305 CrossRef CAS PubMed
- M. Wormuth, M. Scheringer, M. Vollenweider and K. Hungerbühler, Risk Anal., 2006, 26, 803 CrossRef PubMed
- J. Jurewicz and W. Hanke, Int. J. Occup. Med. Environ. Health, 2011, 24, 115 Search PubMed
- European Chemicals Burea, Risk assessment report for bis(2-ethylhexyl)phthalate, D EHP consolidated Final Report. February, 2004. Document No. R042_0402_env_hh_4–6 Search PubMed
- European Food Safety Authority, Statement on the Scientific Panel on Food Additives, 26 May 2004, http://ww.efsa.europa.eu/de/science/scientific_reports/phthalates.html, accessed January 2015 Search PubMed
- CSTEE (Scientific Committee on Toxicity, Ecotoxicity and the Environment), Phthalate migration from soft PVC toys and child-care articles. Opinion expressed at the CSTEE third plenary meeting, Brussels, April 24, 1998 Search PubMed
- CSTEE (Scientific Committee on Toxicity, Ecotoxicity and the Environment), Opinion on phthalate migration from soft PVC toys and child-care articles-Date made available since the 16th of June 1998, opinion expressed at the Sixth CSTEE Plenary Meeting, Brussels, 26/27 November, 1998 Search PubMed
- CSTEE (Scientific Committee on Toxicity, Ecotoxicity and the Environment). Opinion on the results of a second risk assessment of bis(2-ethylhexyl)phthalate (DEHP) in human health part. Adopted by the CSTEE during the 41th Plenary Meeting of 8 January, 2004.
- IARC, IARC Monogr. Eval. Carcinog. Risks Hum., 2000, 77, 41 Search PubMed
- F. Nakane, M. Kunieda, S. Shimizu, Y. Kobayashi, H. Akane, Y. Akie, A. Saito, M. Noguchi, T. Kadota and K. Mitsumori, J. Toxicol. Sci., 2012, 37, 527 CrossRef CAS
- J. H. Petersen and L. K. Shen, Food Addit. Contam., Part A, 2010, 27, 1608 CrossRef CAS PubMed
- K. E. Clark, R. M. David, R. Guinn, K. W. Kramarz, M. A. Lampi and C. A. Staples, Hum. Ecol. Risk Assess., 2011, 17, 923 CrossRef CAS PubMed
- Y. Xu, E. A. Cohen-Hubal and J. C. Little, Environ. Health Perspect., 2010, 118, 253 CAS
- L. E. Gray, J. Ostby, J. Furr, D. N. R. Veeramachaneni and L. Parks, Toxicol. Sci., 2000, 58, 350 CrossRef CAS PubMed
- M. Petrovic, E. Eljarrat, M. J. Lòpez de Alda and D. Barcelò, TrAC, Trends Anal. Chem., 2001, 20, 637 CrossRef CAS
- S. Wang, W. Yang, M. Shi, X. Sun, W. Pang and G. Wang, Chromatographia, 2013, 76, 529 CAS
- X.-L. Cao, Compr. Rev. Food Sci. Food Saf., 2010, 9, 21 CrossRef CAS PubMed
- U.S. F.D.A., Guidance for industry limiting the use of certain phthalates as excipients in CDER-regulated products, Center for Drug Evaluation and Research (CDER), Food and Drug Administration, Silver Spring, USA, December 2012, http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm, accessed January 2015 Search PubMed
- G. Cinelli, P. Avino, I. Notardonato, A. Centola and M. V. Russo, Food Chem., 2014, 146, 181 CrossRef CAS PubMed
- J. Austrian, Environ. Health Perspect., 1973, 4, 3 CrossRef
- W. H. Laurence, Clin. Toxicol., 1978, 13, 89 CrossRef PubMed
- L. Castle, A. J. Mercer, G. R. Startin and J. Gilbert, Food Addit. Contam., 1987, 4, 399 CrossRef CAS PubMed
- E. F. Group Jr, Environ. Health Perspect., 1986, 65, 337 CAS
- R. J. Law, T. W. Fileman and P. Matthiessen, Water Sci. Technol., 1991, 24, 127 CAS
- G. Rock, R. S. Labow and M. Tocchi, Environ. Health Perspect., 1986, 65, 309 CAS
- D. J. Russell, B. McDuffie and S. Fineberg, J. Environ. Sci. Health, Part A: Environ. Sci. Eng., 1985, 20, 927 CrossRef
- M. C. Pietrogrande, D. Rossi and G. Paganetto, Anal. Chim. Acta, 2003, 480, 1 CrossRef CAS
- A. R. Singh, W. H. Lawrence and J. Autian, J. Pharm. Sci., 1972, 61, 51 CrossRef CAS PubMed
- Y. Yagi, Y. Nakamura, I. Tomita, K. Tsuchikawa and N. Shimoi, J. Environ. Pathol. Toxicol., 1980, 4, 533 CAS
- H. A. A. M. Dirven, P. H. H. Van den Broek, J. G. P. Peters, J. Noordhoek and F. J. Jongeneelen, Biochem. Pharmacol., 1992, 43, 2621 CrossRef CAS
- T. Tsutui, E. Watanabe and J. C. Barret, Carcinogenesis, 1993, 14, 611 CrossRef PubMed
- H. A. A. M. Dirven, P. H. H. Van den Broek, M. C. E. Peeters, J. G. Peters, W. C. Mennes, B. J. Blaauboer, J. Noordhoek and F. J. Jongeneelen, Biochem. Pharmacol., 1993, 45, 2425 CrossRef CAS
- G. Paganetto, F. Campi, K. Varani, A. Piffanelli and G. Giovannini, Pharmacol. Toxicol., 2000, 86, 24 CAS
- T. Wenzl, Methods for the determination of phthalates in food, European EUR 23682 EN, Joint Research Centre - Institute for Reference Materials and Measurements; Office for Official Publications of the European Communities, 2009, p. 49, http://www.bezpecnostpotrain.cz/UserFiles/File/Publikace/ftalaty.pdf, ISBN 978-92-79-11125-9, accessed January 2015 Search PubMed
- IUPAC, Compendium of Chemical Terminology, Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997, (the “Gold Book”), XLM on-line corrected version available at http://www.goldbook.iupac.org, updated by M. Nic, J. Jirat, B. Kosata on 2006, ISBN 0-9678550-9-8 Search PubMed
- A. Fankhauser-Noti and K. Grob, Anal. Chim. Acta, 2007, 582, 353 CrossRef CAS PubMed
- G. Prokupkovà, K. Holadovà, J. Poustka and J. Hajslova, Anal. Chim. Acta, 2002, 457, 211 CrossRef
- H.-Y. Shen, Talanta, 2005, 66, 734 CrossRef CAS PubMed
- M. Del Carlo, A. Pepe, G. Sacchetti, D. Compagnone, D. Mastrocola and A. Cichelli, Food Chem., 2008, 111, 771 CrossRef CAS PubMed
- M. V. Russo, I. Notardonato, G. Cinelli and P. Avino, Anal. Bioanal. Chem., 2012, 402, 1373 CrossRef CAS PubMed
- G. Cinelli, P. Avino, I. Notardonato, A. Centola and M. V. Russo, Anal. Chim. Acta, 2013, 769, 2 CrossRef PubMed
- M. V. Russo, I. Notardonato, P. Avino and G. Cinelli, RSC Adv., 2014, 4, 59655 RSC
- M. Marega, K. Grob, S. Moreta and L. Conte, J. Chromatogr. A, 2013, 1273, 105 CrossRef CAS PubMed
- L. Brossa, R. M. Marcé, F. Borrull and E. Pocurull, J. Chromatogr. A, 2002, 963, 287 CrossRef CAS
- F. K. Kawahara and J. W. Hodgeson, EPA Meth. 506, ed. D. J. Munch, US EPA, National Exposure Research Laboratory, Office of Research and Development, US Environmental Protection Agency, Cincinnati, Ohio 45268, 1995, rev. 1.1 Search PubMed
- Z. Guo, S. Wang, D. Wei, M. Wang, H. Zhang, P. Gai and J. Duan, Meat Sci., 2010, 84, 484 CrossRef CAS PubMed
- D. Scalici, G. Presti, S. Giuliano and R. Giacalone, Riv. Ital.
Sostanze Grasse, 2011, LXXXVIII, 24 Search PubMed
- F. J. Schenck and S. J. Lehotay, J. Chromatogr. A, 2000, 868, 51 CrossRef CAS
- F. J. Schenck, S. J. Lehotay and V. Vega, J. Sep. Sci., 2002, 25, 883 CrossRef CAS
- Y. Saito, S. Kodama, A. Matsunaga and A. Yamamoto, J. AOAC Int., 2004, 87, 1356 CAS
- J. Sánchez-Avila, M. Fernandez-Sanjuan, J. Vicente and S. Lacorte, J. Chromatogr. A, 2011, 1218, 6799 CrossRef PubMed
- A. Guart, F. Bono-Blay, A. Borrell and S. Lacorte, Food Addit. Contam., 2011, 28, 676 CrossRef CAS PubMed
- Y. Liu, S. Wang and L. Wang, J. Agric. Food Chem., 2013, 61, 1160 CrossRef CAS PubMed
- M. V. Russo, I. Notardonato, P. Avino and G. Cinelli, Anal. Methods, 2014, 6, 7030 RSC
- P.-G. Wu, X.-D. Pan, B.-J. Ma, L.-Y. Wang and J. Zhang, Eur. Food Res. Technol., 2014, 238, 607 CrossRef CAS
- Y.-Y. Gu, X.-J. Yu, J.-F. Peng, S.-B. Chen, Y.-Y. Zhong, D.-Q. Yin and X.-L. Hu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 965, 164 CrossRef CAS PubMed
- Y.-B. Luo, Q.-W. Yu, B.-F. Yuan and Y.-Q. Feng, Talanta, 2012, 90, 123 CrossRef CAS PubMed
- Q. Ye, L. Liu, Z. Chen and L. Hong, J. Chromatogr. A, 2014, 1329, 24 CrossRef CAS PubMed
- J. Pawliszyn, Solid Phase Microextraction: Theory and Practice, Wiley-VCH, New York, 1997 Search PubMed
- A. Peñalver, E. Pocurull, F. Borrull and R. M. Marcé, Trends Anal. Chem., 1999, 18, 557 CrossRef
- A. Peñalver, E. Pocurull, F. Borrull and R. M. Marcé, J. Chromatogr. A, 1999, 839, 253 CrossRef
- M. Möder, P. Popp and J. Pawliszyn, J. Microcolumn Sep., 1998, 10, 225 CrossRef
- M. T. Kelly and M. Larroque, J. Chromatogr. A, 1999, 841, 177 CrossRef CAS
- K. Luks-betlej, P. Popp, B. Janoszka and H. Paschke, J. Chromatogr. A, 2001, 938, 93 CrossRef CAS
- M. Polo, M. Llompart, C. Garcia-Jares and R. Cela, J. Chromatogr. A, 2005, 1072, 63 CrossRef CAS PubMed
- J. He, R. Lv, H. Zhan, H. Wang, J. Cheng, K. Lu and F. Wang, Anal. Chim. Acta, 2010, 674, 53 CrossRef CAS PubMed
- M. B. Gholivand, M. Khodadadian and F. Ahmadi, Anal. Chim. Acta, 2010, 658, 225 CrossRef CAS PubMed
- S. Scorrano, L. Longo and G. Vasapollo, Anal. Chim. Acta, 2010, 659, 167 CrossRef CAS PubMed
- H. Shaikh, N. Memon, H. Khan, M. I. Bhanger and S. M. Nizamani, J. Chromatogr. A, 2012, 1247, 125 CrossRef CAS PubMed
- H. Asadollahzadeh, E. Noroozian and S. Maghsoudi, Anal. Chim. Acta, 2010, 669, 32 CrossRef CAS PubMed
- M.-R. Lee, F.-Y. Lai, J. Dou, K.-L. Lin and L.-W. Chung, Anal. Lett., 2011, 44, 676 CrossRef CAS
- J. Meng, J. Bu, C. Deng and X. Zhang, J. Chromatogr. A, 2011, 1218, 1585 CrossRef CAS PubMed
- M.-H. Dévier, K. Le Menach, L. Viglino, L. Di Gioia, P. L. Lachassagne and H. Budzinski, Sci. Total Environ., 2013, 443, 621 CrossRef PubMed
- M. H. Banitaba, S. S. H. Davarani and A. Pourahadi, J. Chromatogr. A, 2013, 1283, 1 CrossRef CAS PubMed
- I. Corni, M. P. Ryan and A. R. Boccaccini, J. Eur. Ceram. Soc., 2008, 28, 1353 CrossRef CAS PubMed
- L. Besra, T. Uchikoshi, T. S. Suzuki and Y. Sakka, J. Eur. Ceram. Soc., 2009, 29, 1837 CrossRef CAS PubMed
- J. Santana, C. Giraudi, E. Marengo, E. Robotti, S. Pires, I. Nunes and E. M. Gaspar, Environ. Sci. Pollut. Res., 2014, 21, 1380 CrossRef CAS PubMed
- M. A. Moreira, L. C. André and Z. L. Cardeal, Int. J. Environ. Res. Public Health, 2014, 11, 507 CrossRef PubMed
- Y.-L. Feng, J. Zhu and R. Sensenstein, Anal. Chim. Acta, 2005, 538, 41 CrossRef CAS PubMed
- J. Zhu, S. P. Phillpis, Y.-L. Feng and X. Yang, Environ. Sci. Technol., 2006, 40, 5276 CrossRef CAS
- J. D. Carrillo, C. Salazar, C. Moreta and M. T. Tena, J. Chromatogr. A, 2007, 1164, 248 CrossRef CAS PubMed
- J. D. Carrillo, M. P. Martínez and M. T. Tena, J. Chromatogr. A, 2008, 1181, 125 CrossRef CAS PubMed
- K. Holadová, G. Prokůpková, J. Hajšlová and J. Poustka, Anal. Chim. Acta, 2007, 582, 24 CrossRef PubMed
- J. J. Rios, A. Morales and G. Márquez-Ruiz, Talanta, 2010, 80, 2076 CrossRef CAS PubMed
- I. Al-Saleh, N. Shinwari and A. Aisabbaheen, J. Toxicol. Sci., 2001, 36, 469 CrossRef
- S. Zhao, M. Wu, F. Zhao and B. Zeng, Talanta, 2013, 117, 146 CrossRef CAS PubMed
- Y. Yang, H. Nan, G. Wang, W. Yang and J. Xu, Anal. Lett., 2013, 46, 2001 CrossRef CAS
- A. Sannino, J. AOAC Int., 2010, 93, 315 CAS
- G. Dugo, V. Fotia, V. Lo Turco, R. Maisano, A. G. Potortì, A. Salvo and G. Di Bella, Food Control, 2011, 22, 982 CrossRef CAS PubMed
- S. Joshi, D. Mishra, G. Bisht and K. S. Khetwal, EXCLI J., 2011, 10, 274 Search PubMed
- N. Nanni, K. Fiselier, K. Grob, M. Di Pasquale, L. Fabrizi, P. Aureli and E. Coni, Food Control, 2011, 22, 209 CrossRef CAS PubMed
- M. T. Das, P. Ghosh and I. S. Thakur, Environ. Pollut., 2014, 189, 118 CrossRef CAS PubMed
- J. Zhou, Y. Qi, H. Wu, Q. Diao, F. Tian and Y. Li, J. Sep. Sci., 2014, 37, 650 CrossRef CAS PubMed
- F. Xu, W. Wang, H. Jiang, Z. Wang, Z. Wang, P. Guo, S. Sun and S. Ding, Food Anal. Methods, 2014, 7, 1619 CrossRef
- M. F. Zaater, Y. R. Tahbou and A. N. Al Sayyed, J. Chromatogr. Sci., 2014, 52, 447 CAS
- M. Rezaee, Y. Assadi, M. R. M. Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1 CrossRef CAS PubMed
- S. Berijani, Y. Assadi, M. Anbia, M. R. Milani Hosseini and E. Aghaee, J. Chromatogr. A, 2006, 1123, 1 CrossRef CAS PubMed
- R. R. Kozani, Y. Assadi, F. Shemirani, M. R. M. Hosseini and M. R. Jamali, Talanta, 2007, 72, 387 CrossRef CAS PubMed
- M. A. Farajzadeh, M. Bahram and J. A. Jonsson, Anal. Chim. Acta, 2007, 591, 69 CrossRef CAS PubMed
- L. Farina, E. Boido, F. Carrau and E. Dellacassa, J. Chromatogr. A, 2007, 1157, 46 CrossRef CAS PubMed
- D. Nagaraju and S. D. Huang, J. Chromatogr. A, 2007, 1161, 89 CrossRef CAS PubMed
- H. Yan, B. Liu, J. Du and K. H. Row, Analyst, 2010, 135, 2585 RSC
- Y. Zhang and H. K. Lee, J. Chromatogr. A, 2013, 1274, 28 CrossRef CAS PubMed
- A. Mousa, C. Basheer and A. R. Al-Arfaj, J. Sep. Sci., 2013, 36, 2003 CrossRef CAS PubMed
- M. V. Russo, I. Notardonato, P. Avino and G. Cinelli, RSC Adv., 2014, 4, 59655 RSC
- G. Leng, W. Chen, M. Zhang, F. Huang and Q. Cao, J. Sep. Sci., 2014, 37, 684 CrossRef CAS PubMed
- M. A. Farajzadeh, P. Khorram and A. A. A. Nabil, J. Sep. Sci., 2014, 37, 1177 CrossRef CAS PubMed
- L. Guo and H. K. Lee, J. Chromatogr. A, 2013, 1300, 24 CrossRef CAS PubMed
- E. Yiantzi, E. Psillakis, K. Tyrovola and N. Kalogerakis, Talanta, 2010, 80, 2057 CrossRef CAS PubMed
- X.-Z. Hu, J.-H. Wu and Y.-Q. Feng, J. Chromatogr. A, 2010, 1217, 7010 CrossRef CAS PubMed
- S.-Y. Wei, M.-I. Leong, Y. Li and S.-D. Huang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 1218, 9142 CAS
- M. A. Farajzadeh, S. Sheykhizadeh and P. Khorram, J. Sep. Sci., 2013, 36, 939 CrossRef CAS PubMed
- L. Guo and H. K. Lee, Anal. Chem., 2014, 86, 3743 CrossRef CAS PubMed
- H. Farahani, P. Norouzi, R. Dinarvand and M. Reza Ganjali, J. Chromatogr. A, 2007, 1172, 105 CrossRef CAS PubMed
- H. Farahani, M. Reza Ganjali, R. Dinarvand and P. Norouzi, Talanta, 2008, 76, 718 CrossRef CAS PubMed
- L. Hou and H. K. Lee, J. Chromatogr. A, 2004, 1038, 37 CrossRef CAS PubMed
- M. Gawlik-Jędrysiak, J. Anal. Chem., 2013, 68, 959 CrossRef
- C. Kubwabo, P. E. Rasmussen, X. Fan, I. Kosarac, F. Wu, A. Zidek and S. L. Kuchta, Indoor Air, 2013, 23, 506 CrossRef CAS PubMed
- L. E. Shaw and D. Lee, Ultrason. Sonochem., 2009, 16, 321 CrossRef CAS PubMed
- K. Jaworek and M. Czaplicka, Polimeros, 2013, 23, 718 CAS
- C.-H. Dong, Y.-F. Liu, W.-F. Yang, X.-L. Sun and G.-G. Wang, Anal. Methods, 2013, 5, 4513 RSC
- T. F. Silva, B. G. Soares, S. C. Ferreira and S. Livi, Appl. Clay Sci., 2014, 99, 93 CrossRef CAS PubMed
- I. Aparacio, J. L. Santos and E. Alonso, Anal. Chim. Acta, 2007, 584, 455 CrossRef PubMed
- T. T. H. Pham, R. D. Tyagi, S. K. Brar and R. Y. Surampalli, Chemosphere, 2011, 82, 923 CrossRef CAS PubMed
- H.-Y. Shen, Talanta, 2005, 66, 734 CrossRef CAS PubMed
- Y.-Y. Chao, T.-Y. Chien, T.-H. Kuo, Y.-A. Lu, Y.-H. Shih, S.-Y. Chen and Y.-L. Hunag, Anal. Methods, 2013, 5, 5602 RSC
- Y.-Y. Chao, C.-H. Lee, T.-Y. Chien, Y.-H. Shih, Y.-A. Lu, T.-H. Kuo and Y.-L. Huang, J. Agric. Food Chem., 2013, 61, 8063 CrossRef CAS PubMed
- World Health Organization, Indoor air quality: organic pollutants, EURO Rep. Stud. 111, Berlin, Germany, 1989 Search PubMed
- A. Prieto, O. Telleria, N. Etxebarria, L. A. Fernández, A. Usobiaga and O. Zuloaga, J. Chromatogr. A, 2008, 1214, 1 CrossRef CAS PubMed
- S. S. Hang Ho, J. C. Chow, J. G. Watson, L. P. Ting Ng, Y. Kwok, K. F. Ho and J. Cao, Atmos. Environ., 2011, 45, 1491 CrossRef PubMed
- A. Cecinato, C. Balducci, D. Mastroianni and M. Perilli, Environ. Sci. Pollut. Res., 2012, 19, 1915 CrossRef CAS PubMed
- M. Aragón, F. Borrull and R. M. Marcé, J. Chromatogr. A, 2013, 1303, 76 CrossRef PubMed
- N. Hayeck, S. Gligorovski, I. Poulet and H. Wortham, Talanta, 2014, 122, 63 CrossRef CAS PubMed
- A. Prieto, O. Zuloaga, A. Usobiaga, N. Etxebarria and L. A. Fernández, J. Chromatogr. A, 2007, 1174, 40 CrossRef CAS PubMed
- R. Rodil and M. Moeder, J. Chromatogr. A, 2008, 1178, 9 CrossRef CAS PubMed
- A. Prieto, O. Telleria, N. Etxebarria, L. A. Fernández, A. Usobiaga and O. Zuloaga, J. Chromatogr. A, 2008, 1214, 1 CrossRef CAS PubMed
- G. Prokůpková, K. Holadová, J. Poustka and J. Hajšlová, Anal. Chim. Acta, 2002, 457, 211 CrossRef
- H.-C. Liu, W. Den, S.-F. Chan and K. T. Kin, J. Chromatogr. A, 2008, 1188, 286 CrossRef CAS PubMed
- Y. Lai, Z. Huang, X. Ge, R. Lin and H. Chen, Chin. J. Chromatogr., 2012, 30, 647 CrossRef CAS
- P. A. Clausen, R. L. Lindeberg Bille, T. Nilsson, V. Hansen, B. Svensmark and S. Bøwadte, J. Chromatogr. A, 2003, 986, 179 CrossRef CAS
- C. J. Hines, A. Y. Yau, M. M. Zuniga, J. R. Wells, N. B. Nilsen Hopf and D. E. Camann, J. Environ. Monit., 2010, 12, 491 RSC
- S. Orecchio, R. Indelicato and S. Barreca, Environ. Geochem. Health, 2013, 35, 613 CrossRef CAS PubMed
- A. Shafaghat, Nat. Prod. Commun., 2011, 6, 1739 CAS
- J. Jamilah, A. A. Sharifa and N. R. S. A. Sharifah, World Appl. Sci. J., 2012, 17, 67 CAS
- R. Singh, S. A. Dar and P. Sharma, Res.
J. Med. Plant, 2012, 6, 123 CAS
- T. Mallikadevi, S. Paulsamy, S. Jamuna and K. Karthika, Asian J. Pharm. Clin. Res., 2012, 5, 163 CAS
- T. Suman, K. Chakkaravarthi and R. Elangomathavan, J. Pharm. Technol., 2013, 6, 1173 Search PubMed
- Z. Niu, X. Ye, L. Fang, Q. Xue and Z. Sun, Chin. J. Chromatogr., 2006, 24, 503 CAS
- R. Gibson, M.-J. Wang, E. Padgett and A. J. Beck, Chemosphere, 2005, 61, 1336 CrossRef CAS PubMed
- Q.-Y. Cai, C.-H. Mo, Q.-T. Wu, Q.-Y. Zeng and A. Katsoyiannis, J. Chromatogr. A, 2007, 1143, 207 CrossRef CAS PubMed
- L. Yang, S. Cheng and Z. Wu, Fresenius Environ. Bull., 2009, 18, 2048 CAS
- F. Paune, J. Caixach, I. Espadaler, J. Om and J. Rivera, Water Res., 1998, 32, 3313 CrossRef CAS
- C. Nerín, E. Asensio and C. Jiménez, Anal. Chem., 2002, 74, 5831 CrossRef
| This journal is © The Royal Society of Chemistry 2015 |