
Ground and excited states of naphthalene–water (naphtha–W6) clusters: a computational study†
Divya Sharma and
Martin J. Paterson*
Institute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot Watt University, Edinburgh, EH14 4AS, UK. E-mail: m.j.paterson@hw.ac.uk
First published on 17th March 2015
Abstract
An MP2 and DFT study of the structures of naphthalene–water hexamer clusters has been performed for both the prism and cage forms of the cluster. These have been corrected for zero-point energy and counterpoise effects and show a larger binding energy for the prism structures. These have been analysed in terms of the number and type of hydrogen-bonding interactions present. We have also performed time-dependent DFT computations of the electronic excited states with a variety of functionals. Our results show good agreement between TD-CAM-B3LYP and TD-M06-2X, and predict that the presence of the water cluster red-shifts and lowers the intensity of the primary π → π* transition. This effect is more pronounced in the prism than the cage cluster. We also calculate weaker higher energy features involving charge transfer from the naphthalene to the water cluster.
Introduction
The properties of water clusters (Wn) have been the subject of interest in many theoretical and experimental studies due to their interesting features, and their fundamental importance in many areas of Science such as Physics, Chemistry, Biology, etc. has been well established in the past.1–9 Many theoretical and experimental research groups have carried out extensive research to study the interactions of water clusters with aromatic molecules. The weak hydrogen bonding interactions in such systems plays an important role in determining their physical and chemical properties. Considering one of the simplest systems to replicate such aromatic–water interactions, benzene–water (Bz–Wn) complexes, has been chosen extensively and studied in detail.10–25 Numerous theoretical and experimental studies have been performed on the ground state properties such as binding energies and IR spectra of Bz–Wn clusters, and also on non-covalent interactions such as hydrogen bonding interactions that dominate such systems.2,7,10,14,19,21–24,26–28 Such aromatic–water interactions are also very important in interstellar ices where polycyclic aromatic hydrocarbons (PAHs) interact with water as the most abundant molecule in icy mantles.29Polycyclic aromatic hydrocarbons (PAHs) as a reservoir of cosmic carbon are regarded as key molecules in the astrochemical evolution of the interstellar medium.30,31 The photo-processing of interstellar ices containing PAHs by UV light has been studied and it is observed to play an important role in the formation of many complex organic species.32 Experiments33 have been performed to study photo-processes in interstellar ice analogs i.e., PAH-ice model systems under interstellar icy conditions that corresponds to diffuse regions of molecular clouds in the interstellar medium. In this experiment,33 benzene was chosen as a prototypical PAH compound and photo-processes in model interstellar ices was studied by modeling multilayer films of benzene & water deposited on a sapphire substrate at a temperature of around 80 K. Three distinct photo-desorption mechanisms were investigated in such systems: (i) direct adsorbate-mediated desorption of benzene; (ii) indirect adsorbate-mediated desorption of water; (iii) substrate-mediated desorption of both benzene and water. It is also observed by recent experimental study34 on photon- and electron- induced desorption from laboratory models of interstellar ice grains that photon absorption by benzene can make H2O desorption possible at wavelengths where the photon-absorption cross-section for H2O is negligible.
Recently, we have performed a detailed computational study on ground and excited states of the benzene–water hexamer (Bz–W6) system.35 UV spectral characteristics and vertical excitation energies were calculated to analyze and characterize electronic excitations in this astrophysical and environmentally relevant system. In this present paper we extend this work to truly polycyclic aromatics via naphthalene (C10H8), consisting of linearly fused two benzene rings. Naphthalene has been identified in the interstellar medium (ISM)36–38 and is considered as a complex molecule in the ISM. However, the formation mechanism of naphthalene in the interstellar medium and its derivatives has always been a subject of debate and interest due to ubiquitous presence of PAHs in the ISM. A recent experimental and theoretical study39 has proposed the formation mechanism of naphthalene in the gas phase via a barrierless and exoergic reaction between the phenyl radical (C6H5) and vinylacetylene involving a van der Waals complex and submerged barrier in the entrance channel. It is also suggested that PAH-formation in the interstellar medium can also occur at low temperatures, and more complex PAHs like phenanthrene and anthracene at temperatures down to 10 K can also be formed in cold molecular clouds.39
We have chosen naphthalene as a representative of PAH family, and water clusters to represent the basic features of an interstellar ice surface. Naphthalene and its derivatives are also used in the production of dyes, resin, plastics, lubricants, fuels, etc., and are also considered environmental pollutants.40 Therefore, study of complexes of naphthalene with water clusters is of both astrophysical and environmental relevance.
Many theoretical41–44 and experimental studies45–48 have been performed to study excited state properties of isolated naphthalene. Naphthalene is of lower symmetry than benzene, which allows many transitions that are forbidden in benzene. Excited state properties of naphthalene–water complex systems have not received much attention so far. In the present computational study, the main aim is to study both ground and excited states of the naphtha–W6 cluster using quantum chemical methods. The linear response time-dependent DFT49–52 with range of well-developed DFT functionals is used to study photochemistry and UV spectroscopy of naphtha–W6 clusters to investigate and characterize important electronic transitions in this system.
We have chosen the water hexamer (H2O)6 cluster for our study, the smallest water cluster which allows non-cyclic structures and more three dimensional structures, and also as this is considered as a benchmark system for many computational chemistry studies on water clusters and their effects.5,20,28,53–55 There have been extensive computational and experimental studies on the ground state properties and hydrogen bonding interactions present in the different conformers of the water hexamer.1,3,5,8,28,53–55 Many theoretical studies have established the cage conformer as the minimum energy structure among all other close lying conformers such as prism, book, ring, chair, etc.7,8,20,55 An experimental study by Pate et al. has also established the cage conformer of water hexamer as the global minimum energy structure.54 However, the most recent computational studies have shown the prism as the lowest energy structure, although nearly iso-energetic to the cage structure.1,53 It has been predicted by both theoretical1 and experimental observations54 that cage and prism conformers exist at low temperatures, which are relevant to interstellar conditions. Thus, we choose these two conformers for the water hexamer for our present study i.e., the cage and the prism conformers of the water hexamer (H2O)6 consisting of eight and nine hydrogen bonds, respectively. This also allows direct comparison of our previous computational study of benzene–W6.35
Computational details
The ground state geometries of both the cage and prism conformers of W6 clusters and their respective naphthalene bound W6 clusters i.e., naphtha–W6 clusters were fully optimized using second order Moller–Plesset perturbation theory (MP2), in addition to density functional theory (DFT) with the long range corrected wB97XD functional. MP2 is considered reliable for ground state geometry optimizations by accounting for dynamic electron-correlation effects including dispersion and has been used extensively to study loosely bound hydrogen bonded and dispersion bound complexes.56 wB97XD is one of the most promising long range corrected DFT functionals, i.e., one that also includes an empirical atom–atom dispersion correction to treat systems involving general non-covalent interactions.57,58 The augmented correlation-consistent polarized-valence double zeta basis set (aug-cc-pVDZ) was employed for geometry optimization. Harmonic vibrational frequency calculations were carried out to check the nature of stationary points, and confirmed as minima for all ground state structures except the MP2/aug-cc-pVDZ optimized naphtha–W6 cage structure which is a stationary point on the MP2/aug-cc-pVDZ potential energy surface but has a very small imaginary frequency. The surface is very flat for this geometry and we have taken this stationary point as it is matches the corresponding DFT stationary point which is a minimum.We have used TD-DFT linear response theory,49–52 one of the most popular and widely used electronic structure methods to calculate electronic excitation energies. TD-DFT calculations are performed on both MP2/aug-cc-pVDZ and wB97XD/aug-cc-pVDZ optimized ground state geometries of naphthalene, W6 and naphtha–W6 clusters using three different functionals i.e., B3LYP,59 CAM-B3LYP,60 and M06-2X61 with the aug-cc-pVDZ basis set. We have also calibrated the TD-DFT calculations on MP2 optimized geometries using the larger TZ basis set. M06-2X is a non-local functional with 52% Hartree–Fock exchange, and are often found to perform better than standard hybrid functionals (such as the popular B3LYP) for systems involving non-covalent interactions, and modeling electronic excitation energies to both valence and Rydberg states.61,62 CAM-B3LYP has been specifically designed to model charge-transfer excitations in TD-DFT by “switching on” Hartree–Fock exchange as a function of distance. Thus, the three functionals chosen represent a reasonable variation in characteristics to better understand the transitions in these naphtha–W6 clusters.
CIS(D)63 calculations were also performed with the aug-cc-pVDZ basis set at the MP2 optimized ground state geometries of naphthalene, W6 and naphtha–W6 clusters, in order to compare TD-DFT with a basic wave function approach accounting for some correlation effects on excitation energies.
The Gaussian09 program64 was used for all computations.
Results and discussion
Ground state geometries and energetics
MP2/aug-cc-pVDZ optimized ground state geometries of the prism and cage form of the naphtha–W6 cluster are shown in Fig. 1(a) and (b), respectively. | ||
| Fig. 1 MP2/aug-cc-pVDZ optimized geometries of naphtha–W6 clusters (a) prism conformer (b) cage conformer. wB97XD/aug-cc-pVDZ optimized geometries of naphtha–W6 clusters (c) prism conformer (d) cage conformer. Aromatic centre of both aromatic rings are shown as purple circle. | ||
The binding energy (BE) of the naphtha–W6 cluster was calculated using
| |BE| = (ENaphthalene–w6 − (Ew6 + ENaphthalene)) | (1) |
| MP2 (without ZPE) | MP2 (with ZPE) | wB97XD (without ZPE) | wB97XD (with ZPE) | |
|---|---|---|---|---|
| PRISM | ||||
| ENaphtha–W6 (au) | −842.4050 | −842.1090 | −844.4083 | −844.1052 |
| EW6 (au) | −457.6430 | −457.4930 | −458.5969 | −458.4445 |
| ENaphthalene (au) | −384.7391 | −384.5932 | −385.7946 | −385.6462 |
| BE (kcal mol−1) | 14.33 (7.57) | 14.29 | 10.51 (8.91) | 9.11 |
| CAGE | ||||
| ENaphtha–W6 (au) | −842.3982 | −842.10332 | −844.4045 | −844.1019 |
| EW6 (au) | −457.6426 | −457.4929 | −458.5966 | −458.4442 |
| ENaphthalene (au) | −384.7391 | −384.5932 | −385.7946 | −385.6462 |
| BE (kcal mol−1) | 10.31 (4.97) | 10.82 | 8.33 (7.01) | 7.24 |
It is noticed that BE's of the naphtha–W6 cluster (with zero point energy (ZPE) correction) ranges from 9.11–14.29 kcal mol−1 for the prism conformer between DFT and MP2, whilst this varies between 7.24–10.82 kcal mol−1 for the cage conformer. In both systems it is observed that MP2 calculations give higher binding energies (after ZPE) than wB97XD calculations. It is seen that both cage and prism conformers of the water hexamer cluster essentially retain their shape after interacting with the naphthalene.
At the MP2 optimized geometries, it is found that naphthalene binds to the water hexamer cluster via hydrogen bonding interactions. In the prism conformer, four O–H⋯π hydrogen bonding type interactions are noticed where dangling hydrogen atoms of the water cluster point toward the π electron cloud of the naphthalene ring (see Fig. 1(a)). In the cage conformer, two O–H⋯π hydrogen bonding interactions are noticed where one of the dangling hydrogen atom of the water cluster interacts with the π electron cloud of the naphthalene ring, including two C–H⋯O type hydrogen bonding interactions where an H atom of the naphthalene ring interacts with the dangling oxygen of the water cluster, gives total of four hydrogen bonding interactions (Fig. 1(b)).
Similarly, in the wB97XD optimized geometries, four O–H⋯π hydrogen bonding type interactions are noticed in the prism conformer too (see Fig. 1(c)), while in cage conformer, three O–H⋯π hydrogen bonding type interactions and one C–H⋯O type hydrogen bonding interactions are seen (Fig. 1(d)). However, shorter bond distances for these hydrogen-bonding interactions are noticed in MP2 optimized geometries as compared to wB97XD-optimized geometries. This may result in stronger binding and hence higher binding energies for naphtha–water clusters calculated by MP2 than by wB97XD.
Basis set superposition error (BSSE) was also estimated for both the cage and prism forms of naphtha–W6 clusters using the counterpoise (CP) method and CP-corrected binding enpergies are also listed in Table 1. BSSE error is significantly larger for MP2 calculations than wB97XD, and binding energies are overestimated by MP2 of calculations for both prism and cage conformers. With BSSE correction, it is seen that binding energies are reduced noticeably for both conformers, varying from 7.57–8.91 kcal mol−1 for the prism conformer between DFT and MP2, while relatively lower binding energies with a wider MP2-DFT variation of 4.97–7.01 kcal mol−1 are observed for the cage conformer. Thus, the prism conformer is found to be more stable than the cage conformer for both MP2 and DFT levels of theory.
In order to check the effects of basis set on the binding energies of naphtha–W6 cage and prism clusters, the ground state geometries of both the cage and prism conformers of W6 clusters along with their respective naphthalene bound W6 clusters and isolated naphthalene molecule were again fully optimized with DFT (wB97XD functional) using a larger basis set i.e., Dunning's correlation-consistent aug-cc-pVTZ basis set. Without ZPE correction, it is noticed that binding energies of naphtha–W6 cluster are 9.41 kcal mol−1 and 7.55 kcal mol−1 for prism and cage conformers, respectively, which differ by about 1 kcal mol−1 compared to respective binding energies calculated using the aug-cc-pVDZ basis set. After BSSE correction the binding energies of naphtha–W6 cluster calculated using aug-cc-pVTZ basis set are 8.99 kcal mol−1 and 7.16 kcal mol−1 for prism and cage conformers, respectively, and are in good agreement with results obtained using aug-cc-pVDZ basis set i.e., within 0.15 kcal mol−1 difference, and hold for both prism and cage conformers. Thus, we can be confident that the smaller basis is accurately describing the binding in these systems.
Electronic excitations in naphthalene bound water W6 clusters
The UV spectral results obtained from TD-DFT calculations on MP2 optimized ground state of the naphtha–W6 cluster, using all three functionals are presented in Fig. 2(a) and 3(a), for the prism and cage conformers respectively. For the prism conformer of the naphtha–W6 cluster, it is noticed that the B3LYP functional generates the strongest peak at around 220–221 nm, while both M06-2X and CAM-B3LYP functionals give the consistent results, and generate the strongest intensity peak at around 212–213 nm (see Fig. 2(a)). Similar trends are noticed for the cage conformer, with UV spectra generated using B3LYP being red-shifted towards compared to M06-2X and CAM-B3LYP functionals, as shown in Fig. 3(a). | ||
| Fig. 2 Simulated UV spectra obtained from TD-DFT calculations on MP2 optimized ground state geometries, (a) comparison of B3LYP, CAM-B3LYP and M06-2X functionals on naphtha–W6 prism cluster. Performance of (b) B3LYP, (c) CAM-B3LYP, and (d) M06-2X on prism shaped water W6 cluster, naphtha–W6 cluster and naphthalene. | ||
 | ||
| Fig. 3 Simulated UV spectra obtained from TD-DFT calculations on MP2 optimized ground state geometries, (a) comparison of B3LYP, CAM-B3LYP and M06-2X functionals on naphtha–W6 cage cluster. Performance of (b) B3LYP, (c) CAM-B3LYP, and (d) M06-2X on cage shaped water W6 cluster, naphtha–W6 cluster and naphthalene. | ||
The effects of the naphthalene interaction with the water cluster W6 are investigated by comparing to those of the isolated water cluster W6, and the isolated naphthalene. The results obtained using all three functionals on the W6 cluster, naphtha–W6 clusters and naphthalene molecule are shown in Fig. 2(b)–(d) for the prism conformer, and Fig. 3(b)–(d) for the cage conformer. The nature of the electronic transitions are analysed by looking at the orbitals involved in the response eigenvectors in those excitations that are associated with higher oscillator strengths or strong intensity peaks in naphtha–W6 spectra, and the important electronic transitions assigned for those excitations are listed in Table 2 and 3 for prism and cage conformer, respectively.
| DFT functional | E (eV) | λ (nm) | Oscillator strength (f) | Character |
|---|---|---|---|---|
| a Exptl/taken from ref. 47.b Exptl/taken from ref. 45.c Exptl/taken from ref. 48.d Exptl/taken from ref. 46. | ||||
| B3LYP | 4.18 (4.22, 4.66a, 4.38b, 4.45c) | 296.5 (294.2) | 0.0483 (0.0547, 0.109c, 0.102d) | π → π* |
| 4.32 (4.33, 4.13a, 4.03b, 4.0c) | 286.9 (286.5) | 0.0007 (0.0000, 0.002d) | π → π* | |
| 5.63 (5.73, 5.62b, 5.89c) | 220.1 (216.3) | 0.6459 (1.2450, 1.3cd) | π → π* | |
| 5.61 | 221.2 | 0.1590 | π → π*(diffuse) | |
| 5.73 | 216.5 | 0.1461 | π → π* | |
| 5.92 | 209.3 | 0.1391 | π → π* | |
| 4.18 | 296.5 | 0.0483 | π → π* | |
| 6.04 | 205.2 | 0.0192 | Naphtha-diffuse state | |
| 6.06 | 204.5 | 0.0191 | Naphtha-CT state | |
| 6.37 | 194.8 | 0.0123 | Water-CT state | |
| CAM-B3LYP | 4.45 (4.48, 4.13a, 4.03b, 4.0c) | 278.4 (277.1) | 0.0338 (0.0000, 0.002d) | π → π* |
| 4.48 (4.50, 4.66a, 4.38b, 4.45c) | 276.6 (275.5) | 0.0295 (0.0708, 0.109c, 0.102d) | π → π* | |
| 5.84 (5.91, 5.62b, 5.89c) | 212.2 (209.8) | 0.7849 (1.1319, 1.3cd) | π → π* | |
| 6.17 | 200.8 | 0.2066 | π → π* | |
| 5.76 | 215.3 | 0.1528 | π → π* | |
| 5.91 | 209.9 | 0.1328 | π → π* (diffuse) | |
| 4.45 | 278.4 | 0.0338 | π → π* | |
| 6.62 | 187.2 | 0.0290 | Naphtha-diffuse state | |
| 6.87 | 180.6 | 0.0157 | Naphtha-CT state | |
| 6.87 | 180.4 | 0.0012 | Water-CT state | |
| 7.21 | 172.1 | 0.0030 | Water-CT state | |
| 7.11 | 174.4 | 0.0091 | Naphtha-CT state | |
| M06-2X | 4.50 (4.52, 4.13a, 4.03b, 4.0c) | 275.3 (274.1) | 0.0149 (0.0000, 0.002d) | π → π* |
| 4.54 (4.57, 4.66a, 4.38b, 4.45c) | 273.3 (271.3) | 0.0510 (0.0737, 0.109c, 0.102d) | π → π* | |
| 5.82 (5.90, 5.62b, 5.89c) | 213.2 (210.2) | 0.8315 (1.2728, 1.3cd) | π → π* | |
| 6.23 | 198.9 | 0.2052 | π → π* | |
| 5.87 | 211.2 | 0.1850 | π → π* | |
| 4.54 | 273.3 | 0.0510 | π → π* | |
| 6.50 | 190.8 | 0.0205 | Naphtha-diffuse state | |
| 6.65 | 186.5 | 0.0119 | Naphtha-CT state | |
| 6.87 | 180.5 | 0.0085 | Naphtha-CT state | |
| 6.90 | 177.3 | 0.0136 | Naphtha-CT state | |
| 6.68 | 185.6 | 0.0001 | Water-CT state | |
| DFT functional | E (eV) | λ (nm) | Oscillator strength (f) | Electronic transition |
|---|---|---|---|---|
| a Exptl/taken from ref. 47.b Exptl/taken from ref. 45.c Exptl/taken from ref. 48.d Exptl/taken from ref. 46. | ||||
| B3LYP | 4.22 (4.22, 4.66a, 4.38b, 4.45c) | 294.7 (294.2) | 0.0499 (0.0547, 0.109c, 0.102d) | π → π* |
| 4.32 (4.33, 4.13a, 4.03b, 4.0c) | 287.2 (286.5) | 0.0008 (0.0000, 0.002d) | π → π* | |
| 5.66 (5.73, 5.62b, 5.89c) | 219.2 (216.3) | 0.7383 (1.2450, 1.3cd) | π → π* | |
| 5.75 | 215.7 | 0.2325 | Naphtha-CT state | |
| 5.91 | 209.9 | 0.1512 | π → π* | |
| 5.89 | 210.6 | 0.0377 | Naphtha-diffuse state | |
| 5.72 | 216.9 | 0.0214 | Naphtha-CT state | |
| 6.13 | 202.2 | 0.0167 | Naphtha-CT state | |
| 6.26 | 198.2 | 0.0089 | Water-CT state | |
| CAM-B3LYP | 4.46 (4.48, 4.13a, 4.03b, 4.0c) | 277.8 (277.1) | 0.0067 (0.0000, 0.002d) | π → π* |
| 4.50 (4.50, 4.66a, 4.38b, 4.45c) | 275.6 (275.5) | 0.0588 (0.0708, 0.109c, 0.102d) | π → π* | |
| 5.87 (5.91, 5.62b, 5.89c) | 211.2 (209.8) | 1.0993 (1.1319, 1.3cd) | π → π* | |
| 6.14 | 201.8 | 0.2189 | π → π* | |
| 5.82 | 212.9 | 0.0375 | π → π* (diffuse) | |
| 6.40 | 193.7 | 0.0216 | Naphtha-CT state | |
| 6.99 | 177.5 | 0.0131 | Naphtha-CT state | |
| 6.93 | 179.0 | 0.0010 | Water-CT state | |
| M06-2X | 4.51 (4.52, 4.13a, 4.03b, 4.0c) | 274.9 (274.1) | 0.0042 (0.0000, 0.002d) | π → π* |
| 4.56 (4.57, 4.66a, 4.38b, 4.45c) | 271.6 (271.3) | 0.0645 (0.0737, 0.109c, 0.102d) | π → π* | |
| 5.85 (5.90, 5.62b, 5.89c) | 211.9 (210.2) | 0.8498 (1.2728, 1.3cd) | π → π* | |
| 5.88 | 210.9 | 0.1727 | π → π* | |
| 6.20 | 199.9 | 0.1974 | π → π* | |
| 5.83 | 212.6 | 0.0500 | Naphtha-diffuse state | |
| 6.26 | 198.2 | 0.0407 | Naphtha-CT state | |
| 6.73 | 184.2 | 0.0002 | Water-CT state | |
Experimentally three absorption bands labeled as α, p, and β are observed for naphthalene, where the p band is polarized along the short axis of the naphthalene and the α and β bands are polarized along the long axis.45 It is observed that the β band is of the strongest intensity; followed by the p and α bands. It is observed that the state corresponding to the α band (polarization along the long axis) is the lowest lying excited state in the naphthalene. For our TD-DFT calculations, we find that the α and the β absorption bands are related to electronic excitations from the ground state to the excited states with symmetry 11B2U and 21B2U, respectively, while p band relates to the electronic excitation to the 11B1U state. All three electronic transitions are assigned as the π → π* transitions. The TD-DFT calculations on naphthalene give consistent results using CAM-B3LYP and M06-2X, and the two lowest lying singlet vertical excitations in naphthalene are of 1B2U symmetry and 1B1U symmetry. This energy ordering is reversed for B3LYP. Both CAM-B3LYP and M06-2X results are in good agreement with the experiment. For the M062X functional, the calculated valence π → π* excitation energies of naphthalene (experimental values in the parenthesis) are 11B2U: 4.52(4.13,47 4.03,45 4.0 (ref. 48)), 11B1U: 4.57(4.66,47 4.38,45 4.45 (ref. 48)), and 21B2U: 5.90(5.7,45 5.89 (ref. 48)) eV, respectively. The α excitation is over-estimated by 0.5 eV for the 11B2U state with the experiments, while the p excitation is calculated with an accuracy of about 0.09 eV for the 11B1U state. The β excitation (i.e., 21B2U state) is described well and is computed with an accuracy of around 0.01 eV. We calculate that the lowest valence transition of naphthalene from ground to excited state 11B2U is a dark state of zero oscillator strength, although not forbidden by symmetry. The naphthalene π → π* electronic transition at around 210 nm (or about 5.9 eV) is of very high intensity i.e., a bright state and corresponds to β absorption band with electronic transition from ground to 21B2U excited state. We have calculated the highest oscillator strength f ≈ 1.3 for 21B2U excited state, which is in good agreement with the experimental observations.
Different excitation features in naphtha–W6 prism cluster are obtained with respect to excitation features in isolated W6 prism cluster and naphthalene molecule by B3LYP calculations, as shown in Fig. 2(b). The naphtha–W6 spectra are red shifted with respect to W6 spectra. The electronic transitions in naphtha–W6 cluster are observed towards longer wavelengths i.e., in the spectral range of about 200–295 nm, whereas in W6 cluster electronic transitions are seen at wavelengths below 176 nm. The naphthalene excitation feature at around 216 nm is of very high intensity, with other less intense peaks at around 208 nm and 294 nm. In the naphtha–W6 prism cluster, the strongest intensity peak at around 220 nm is due to the π → π* electronic transition of naphthalene. Another intense peaks at around 217 nm and 209 nm are also associated with the naphthalene π → π* electronic transition. Some weak electronic transitions at around 204–205 nm correspond to a naphthalene locally diffuse state and its charge transfer (CT) excitation to the water W6 cluster. It is also interesting to observe a weak charge transfer transition caused by water excitations in naphtha–W6 cluster at around 195 nm, which are completely absent in the isolated W6 cluster.
The CAM-B3LYP functional predicts a strongest intensity peak at around 212 nm in the naphtha–W6 cluster which is in close proximity to the naphthalene excitation feature at 210 nm (see Fig. 2(c)). The is the main π → π* transition. All strong intensity peaks found at around 201 nm, 215 nm, and 210 nm are also assigned as π → π* electronic transitions of naphthalene. Some weak transitions at around 278 nm and 187 nm are assigned as a π → π* electronic transition of naphthalene and a locally diffuse state of naphthalene, respectively. A few very weak transitions due to water CT excitations and naphthalene CT excitations are observed at around 180 nm in naphtha–W6 cluster.
Similar UV spectral characteristics are predicted by M06-2X functional, as shown in Fig. 2(d). In naphtha–W6 cluster, the strongest peak at around 213 nm is associated with the π → π* transition feature of naphthalene, while other strong peaks at around 199 nm, and 211 nm are also due to π → π* electronic transitions of naphthalene. A few weak intensity transitions at 273 nm and 191 nm correspond to a π → π* electronic transition of naphthalene and naphthalene locally diffuse state, respectively. Naphthalene weak CT excitations to the water cluster are also observed around 180–185 nm, including small contributions from very weak charge transfer excitations of the water cluster to naphthalene too.
For all functionals we see that the strongest intensity naphthalene π → π* electronic transition in naphtha–W6 prism clusters are red-shifted as compared to that in isolated naphthalene. New weaker, non-additive features such as inter-CT states appear at higher energies for all functionals. Given the nature of these the CAM-B3LYP results are probably the most accurate.
Next, considering the cage conformer of the W6 cluster interacting with naphthalene i.e., naphtha–W6 cage shaped cluster, we have obtained results consistent with those presented above for the prism cluster one. The B3LYP functional predicts the π → π* transition feature of naphthalene at around 219 nm as the strongest transition (see Fig. 3(b)), while both CAM-B3LYP and M06-2X functionals generate the this at around 211 nm (see Fig. 3(c) and (d)). All other important electronic transitions found in naphtha–W6 cage shaped cluster are given in Table 3. It is found that most of the strong intensity electronic transitions in both naphtha–W6 prism and cage clusters are influenced by naphthalene excitations as compared to water W6 excitations. Similarly, naphthalene charge transfer states to cage-shaped water cluster are also observed, showing the effect of naphthalene to enhance water excitations towards longer wavelengths. The presence of weak water charge transfer excitations in these cage shaped naphtha–W6 are also observed. It is also noted that oscillator strengths of the strongest transition are much higher in naphthalene and naphtha–W6 cluster than those in W6 cluster, which hold for all three functionals. It is also calculated that the cage naphtha–W6 cluster shows higher individual transition oscillator strengths for bright states than the prism naphtha–W6 cluster (see Tables 2 and 3). Fig. 4 illustrates the nature of the molecular orbitals involved in the electronic transitions for some of the electronic excited states computed at the TD-DFT level for both cage and prism conformers of naphtha–W6 clusters.
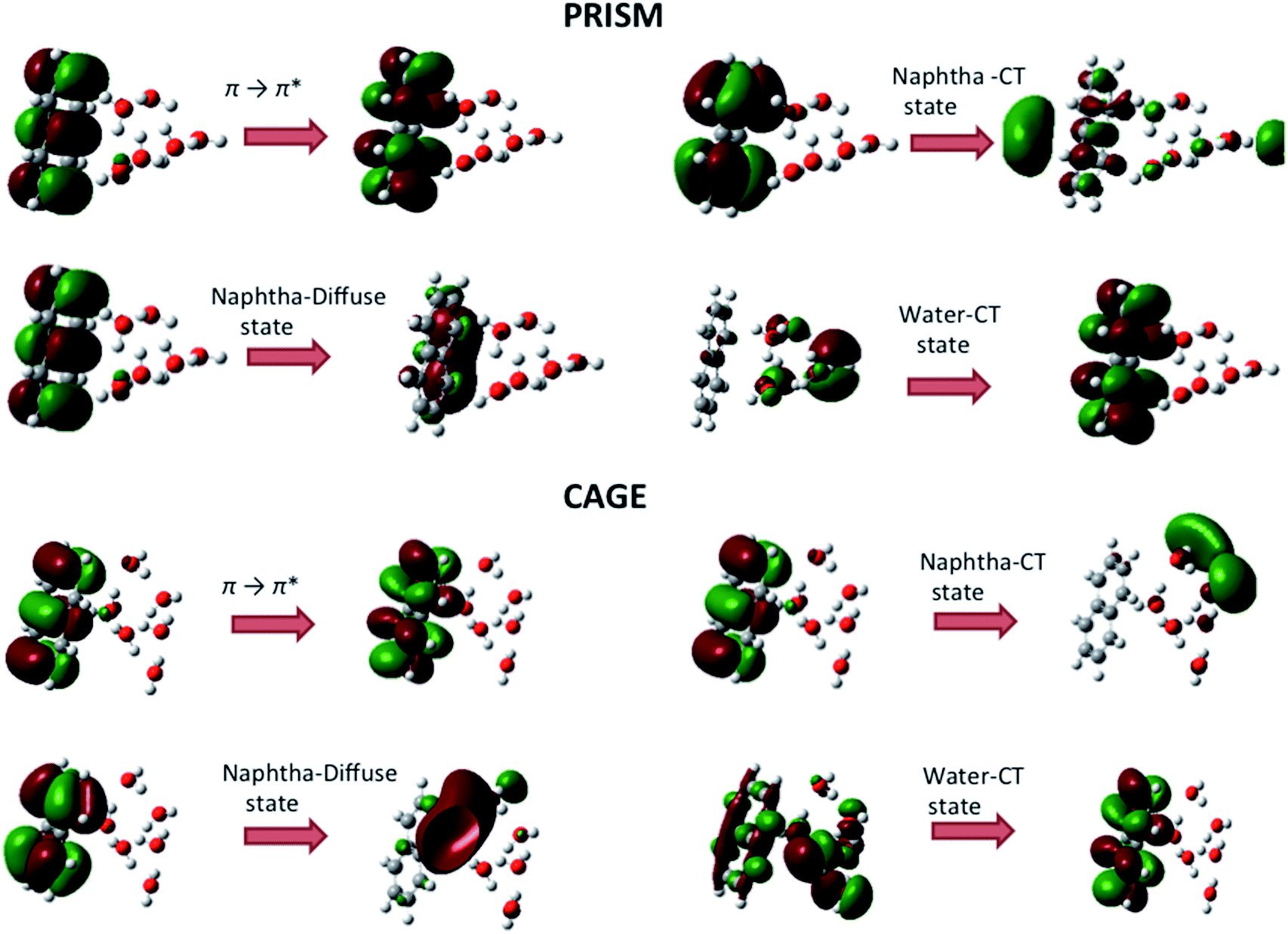 | ||
| Fig. 4 Molecular orbitals involved in the electronic transitions for few electronic excited states computed at the TD-DFT level for both prism and cage conformers of naphtha–W6 cluster. | ||
In order to calibrate basis set effects on the UV spectra, we have also performed TD-DFT calculations using larger basis set at MP2 optimized ground state geometries of naphthalene, W6 and naphtha–W6 clusters. For all three functionals, it is noticed that electronic excitations show only a small red-shift <0.04 eV for both isolated naphthalene and naphtha–W6 clusters relative to those electronic excitations generated using aug-cc-pVDZ basis set, while a small blue shift of <0.07 eV is seen in W6 clusters for both prism and cage conformers. The nature of excitations associated with higher oscillator strengths in naphtha–W6 clusters (for prism and cage conformers) is generally quite consistent for both aug-cc-pVDZ and aug-cc-pVTZ calculations (see ESI: Tables S1 and S2†).
We have also compared the TD-DFT excitations with a correlated approach by performing CIS(D) calculations i.e., configuration interaction with single excitations and added doubles (D) perturbation on those, in conjunction with the aug-cc-pVDZ basis set, on the MP2 optimized ground state geometries of naphthalene, W6 and naphtha–W6 clusters. It is noticed that the lowest dark excited state of naphthalene shows a red-shift of about 0.10–0.15 eV, while the brightest excited state shows a blue-shift of about 0.15 eV with respect to those in TD-CAM and TD-M062X results. We observe similar effects in the naphtha–W6 cage and prism conformers, and the lowest excited state of very small oscillator strength gives a red-shift of about 0.15 eV, while the brightest state gives a blue-shift of about 0.30 eV with respect to those in TD-CAM and the TD-M062X results. The largest discrepancy seen is for a state where the CIS(D) is very similar to the (uncorrelated) CIS and gives a blue-shift of about 0.5–0.6 eV with respect to those in TD-DFT results.
Finally, UV spectra results obtained at wB97XD/aug-cc-pVDZ optimized geometries are given in ESI.† For the prism and the cage conformer of the naphtha–W6 cluster, UV spectra are given in Fig. S1 and S2 (see ESI†), respectively and their corresponding important electronic transitions are also listed in Tables S1 and S2 (see ESI†), respectively. Very similar UV spectral characteristics are observed for the wB97XD optimized naphtha–W6 prism and cage shaped cluster, where UV spectra undergo a red shift in going from the CAM-B3LYP to the B3LYP functional (see ESI: Fig. S1 and S2 and Tables S3 and S4†). We observe stronger peak intensities in naphtha–W6 cluster than those in the isolated W6 cluster. It is again noted that the presence of naphthalene enhances the excitations in W6 cluster towards wavelengths above 170 nm, therefore generating new naphthalene-CT and water-CT states too. Again for all three functionals, it is found that the strongest intensity peak (bright state) associated with the naphthalene π → π* transition undergoes a small red shift in naphtha–W6 cluster with respect to those in isolated naphthalene molecule. It is calculated that in naphtha–W6 cluster, the strongest intensity transition (bright state) of the naphthalene shows a blue-shift of around 4–5 nm for wB97XD optimized geometries with respect to those in MP2 optimized geometries, and hold for both prism and cage conformers.
Conclusions
We have performed both DFT-wB97XD and MP2 calculations to obtain optimized ground state geometries of naphthalene, water hexamer W6 and naphthalene bound W6 clusters. We show that naphthalene binds more strongly to the prism conformer of the water hexamer cluster than the cage conformer as binding energies are calculated higher for the prism conformer than the cage conformer. In both naphthalene-bound prism and cage shaped W6 clusters, O–H⋯π type hydrogen bonding interactions are found to dominate and provide stability in these clusters, while some contribution from C–H⋯O type hydrogen bonding interactions are also present in the cage conformer.Further we have generated vertical excited states by performing linear response time-dependent DFT calculations on both MP2 and wB97XD optimized geometries of naphthalene, water hexamer W6 and naphthalene-bound W6 clusters. UV spectral characteristics are noticeably different for both cage and prism conformers of W6 and naphtha–W6 clusters. It is found that TD-DFT results obtained using CAM-B3LYP and M06-2X functionals are in good agreement. The π → π* electronic transitions of naphthalene show a small red-shift in naphtha–W6 cluster (for both cage and prism conformers) with respect to those in isolated naphthalene. The intensities of the π → π* naphthalene excitations are found to be lower in naphtha–W6 cluster relative to those in isolated naphthalene due to the presence of water cluster around it.
We have shown some interesting features of naphthalene-mediated water cluster excitations at wavelengths above 170 nm in both naphthalene-bound W6 cage and prism water clusters which are not seen in isolated water W6 clusters, indicating the effect of naphthalene on water excitations. Our results are in good agreement with the recent experimental33,34,65 and computational studies,38 where it was shown that benzene as a prototypical PAH molecule can act as a mediator to excite water molecules and shift water excitations to lower energy, where the photon-absorption cross-section for water is negligible at such wavelengths. A similar effect is noticed in naphthalene-bound water hexamer clusters, and is more pronounced than benzene-bound water clusters. Our results also predict new naphthalene charge transfer (CT) states and locally excited diffuse states which also influence water excitations in such naphthalene-bound water complexes.
Acknowledgements
Thanks are given to Prof. Martin McCoustra for helpful discussion. DS acknowledges the LASSIE FP7 Marie Curie Initial Training Network (ITN) for funding, while both DS and MJP acknowledge support from European Research Council (ERC grant number 25899).References
- D. M. Bates and G. S. Tschumper, J. Phys. Chem. A, 2009, 113, 3555–3559 CrossRef CAS PubMed.
- A. Lenz and L. Ojamae, Phys. Chem. Chem. Phys., 2005, 7, 1905–1911 RSC.
- A. Lenz and L. Ojamae, J. Phys. Chem. A, 2006, 110, 13388–13393 CrossRef CAS PubMed.
- A. Lenz and L. Ojamae, Chem. Phys. Lett., 2006, 418, 361–367 CrossRef CAS.
- K. Liu, M. G. Brown, C. Carter, R. J. Saykally, J. K. Gregory and D. C. Clary, Nature, 1996, 381, 501–503 CrossRef CAS.
- S. Maheshwary, N. Patel, N. Sathyamurthy, A. D. Kulkarni and S. R. Gadre, J. Phys. Chem. A, 2001, 105, 10525–10537 CrossRef CAS.
- Y. I. Neela, A. S. Mahadevi and G. N. Sastry, J. Phys. Chem. B, 2010, 114, 17162–17171 CrossRef CAS PubMed.
- D. M. Upadhyay, M. K. Shukla and P. C. Mishra, Int. J. Quantum Chem., 2001, 81, 90–104 CrossRef CAS.
- Y. Wang, V. Babin, J. M. Bowman and F. Paesani, J. Am. Chem. Soc., 2012, 134, 11116–11119 CrossRef CAS PubMed.
- M. Alberti, N. F. Lago and F. Pirani, Chem. Phys., 2012, 399, 232–239 CrossRef CAS.
- M. Baron and V. J. Kowalewski, J. Phys. Chem. A, 2006, 110, 7122–7129 CrossRef CAS PubMed.
- D. Feller, J. Phys. Chem. A, 1999, 103, 7558–7561 CrossRef CAS.
- S. Y. Fredericks, J. M. Pedulla, K. D. Jordan and T. S. Zwier, Theor. Chem. Acc., 1997, 96, 51–55 CrossRef CAS.
- A. W. Garrett and T. S. Zwier, J. Chem. Phys., 1992, 96, 3402–3410 CrossRef CAS.
- A. J. Gotch and T. S. Zwier, J. Chem. Phys., 1992, 96, 3388–3401 CrossRef CAS.
- C. J. Gruenloh, J. R. Carney, C. A. Arrington, T. S. Zwier, S. Y. Fredericks and K. D. Jordan, Science, 1997, 276, 1678–1681 CrossRef CAS.
- C. J. Gruenloh, J. R. Carney, F. C. Hagemeister, C. A. Arrington, T. S. Zwier, S. Y. Fredericks, J. T. Wood and K. D. Jordan, J. Chem. Phys., 1998, 109, 6601–6614 CrossRef CAS.
- P. Jedlovszky, A. Kereszturi and G. Horvai, Faraday Discuss., 2005, 129, 35–46 RSC.
- S. J. Kim, H. I. Seo and B. H. Boo, Mol. Phys., 2009, 107, 1261–1270 CrossRef CAS.
- J. Y. Lee, J. Kim, H. M. Lee, P. Tarakeshwar and K. S. Kim, J. Chem. Phys., 2000, 113, 6160–6168 CrossRef CAS.
- M. Prakash, K. G. Samy and V. Subramanian, J. Phys. Chem. A, 2009, 113, 13845–13852 CrossRef CAS PubMed.
- R. N. Pribble, A. W. Garrett, K. Haber and T. S. Zwier, J. Chem. Phys., 1995, 103, 531–544 CrossRef CAS.
- R. N. Pribble and T. S. Zwier, Science, 1994, 265, 75–79 CAS.
- R. N. Pribble and T. S. Zwier, Faraday Discuss., 1994, 97, 229–241 RSC.
- P. Tarakeshwar, H. S. Choi, S. J. Lee, J. Y. Lee, K. S. Kim, T. K. Ha, J. H. Jang, J. G. Lee and H. Lee, J. Chem. Phys., 1999, 111, 5838–5850 CrossRef CAS.
- J. Ma, D. Alfe, A. Michaelides and E. Wang, J. Chem. Phys., 2009, 130 CAS.
- R. N. Pribble and T. S. Zwier, Science, 1994, 265, 75–79 CAS.
- K. Kim, K. D. Jordan and T. S. Zwier, J. Am. Chem. Soc., 1994, 116, 11568–11569 CrossRef CAS.
- L. J. Allamandola, M. P. Bernstein, S. A. Sandford and R. L. Walker, Space Sci. Rev., 1999, 90, 219–232 CrossRef CAS PubMed.
- L. J. Allamandola, Abstracts of Papers of the American Chemical Society, 2006, 231 Search PubMed.
- L. J. Allamandola, D. M. Hudgins, C. W. Bauschlicher and S. R. Langhoff, Astron. Astrophys., 1999, 352, 659–664 CAS.
- M. P. Bernstein, S. A. Sandford, L. J. Allamandola, J. S. Gillette, S. J. Clemett and R. N. Zare, Science, 1999, 283, 1135–1138 CrossRef CAS PubMed.
- J. D. Thrower, M. P. Collings, M. R. S. McCoustra, D. J. Burke, W. A. Brown, A. Dawes, P. D. Holtom, P. Kendall, N. J. Mason, F. Jamme, H. J. Fraser, I. P. Clark and A. W. Parker, J. Vac. Sci. Technol., A, 2008, 26, 919–924 CAS.
- J. D. Thrower, A. G. M. Abdulgalil, M. P. Collings, M. R. S. McCoustra, D. J. Burke, W. A. Brown, A. Dawes, P. J. Holtom, P. Kendall, N. J. Mason, F. Jamme, H. J. Fraser and F. J. M. Rutten, J. Vac. Sci. Technol., A, 2010, 28, 799–806 CAS.
- D. Sharma and M. J. Paterson, Photochem. Photobiol. Sci., 2014, 13, 1549–1560 CAS.
- S. Iglesias-Groth, A. Manchado, D. A. Garcia-Hernandez, J. I. G. Hernandez and D. L. Lambert, Astrophys. J., Lett., 2008, 685, L55–L58 CrossRef CAS.
- J. Krelowski, G. A. Galazutdinov, F. A. Musaev and J. Nirski, Mon. Not. R. Astron. Soc., 2001, 328, 810–814 CrossRef.
- T. P. Snow, Astrophys. J., 1992, 401, 775–777 CrossRef CAS.
- D. S. N. Parker, F. T. Zhang, Y. S. Kim, R. I. Kaiser, A. Landera, V. V. Kislov, A. M. Mebel and A. G. G. M. Tielens, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 53–58 CrossRef CAS PubMed.
- R. Preuss, J. Angerer and H. Drexler, Int. Arch. Occup. Environ. Health, 2003, 76, 556–576 CrossRef CAS PubMed.
- T. Hashimoto, H. Nakano and K. Hirao, J. Chem. Phys., 1996, 104, 6244–6258 CrossRef CAS.
- E. S. Kadantsev, M. J. Stott and A. Rubio, J. Chem. Phys., 2006, 124 Search PubMed.
- K. Lopata, R. Reslan, M. Kowaska, D. Neuhauser, N. Govind and K. Kowalski, J. Chem. Theory Comput., 2011, 7, 3686–3693 CrossRef CAS.
- M. J. Packer, E. K. Dalskov, T. Enevoldsen, H. J. A. Jensen and J. Oddershede, J. Chem. Phys., 1996, 105, 5886–5900 CrossRef CAS.
- D. Biermann and W. Schmidt, J. Am. Chem. Soc., 1980, 102, 3163–3173 CrossRef CAS.
- G. A. George and G. C. Morris, J. Mol. Spectrosc., 1968, 26, 67–71 CrossRef CAS.
- S. Grimme and M. Parac, ChemPhysChem, 2003, 4, 292–295 CrossRef CAS PubMed.
- R. H. Huebner, S. R. Mielczar and C. E. Kuyatt, Chem. Phys. Lett., 1972, 16, 464–469 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- K. Burke, J. Werschnik and E. K. U. Gross, J. Chem. Phys., 2005, 123 Search PubMed.
- M. L. Marques and E. K. U. Gross, Annu. Rev. Phys. Chem., 2004, 55, 427–455 CrossRef CAS PubMed.
- B. Temelso, K. A. Archer and G. C. Shields, J. Phys. Chem. A, 2011, 115, 12034–12046 CrossRef CAS PubMed.
- C. Perez, M. T. Muckle, D. P. Zaleski, N. A. Seifert, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Science, 2012, 336, 897–901 CrossRef CAS PubMed.
- M. D. Tissandier, S. J. Singer and J. V. Coe, J. Phys. Chem. A, 2000, 104, 752–757 CrossRef CAS.
- K. E. Riley and P. Hobza, J. Phys. Chem. A, 2007, 111, 8257–8263 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef.
- M. Headgordon, R. J. Rico, M. Oumi and T. J. Lee, Chem. Phys. Lett., 1994, 219, 21–29 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. L. Caricato, X. H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. P. Cammi, C. J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- J. D. Thrower, D. J. Burke, M. P. Collings, A. Dawes, P. D. Holtom, F. Jamme, P. Kendall, W. A. Brown, I. P. Clark, H. J. Fraser, M. R. S. McCoustra, N. J. Mason and A. W. Parker, Astrophys. J., 2008, 673, 1233–1239 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electronic transitions obtained using B3LYP, CAM-B3LYP and M06-2X hybrid functionals with aug-cc-pVTZ basis set on MP2 optimized naphtha–W6 prism and cage shaped clusters (Tables S1 and S2); UV spectra (Fig. S1 and S2) and important electronic transitions corresponding to higher oscillator strengths for wB97XD optimized naphtha–W6 prism and cage shaped clusters (Tables S3 and S4). See DOI: 10.1039/c5ra01894c |
| This journal is © The Royal Society of Chemistry 2015 |