DOI:
10.1039/C5RA01883H
(Paper)
RSC Adv., 2015,
5, 34439-34446
Theoretical investigation of the mechanism of gold(I)-catalyzed hydrothiolation of alkynes and alkenes with phenthiol†
Received
30th January 2015
, Accepted 8th April 2015
First published on 8th April 2015
Abstract
The mechanisms of the gold-catalyzed hydrothiolation of alkynes and alkenes with phenthiol have been investigated using density functional theory calculations done at the B3LYP/6-31G (d,p) (SDD for Au) level of theory. The solvent effect was taken into account by B3LYP/6-311++G (d,p) (SDD for Au) single-point calculations with the integral equation formalism polarizable continuum model (IEFPCM) and solvation model (SMD) in toluene. The calculations indicated that the reactions of the gold-catalyzed hydrothiolation proceed through two competing pathways and lead to Markovnikov-type sulfides or anti-Markovnikov-type products. The process of forming anti-Markovnikov-type products is more favored kinetically with barriers of 21.9 and 23.6 kcal mol−1 for alkyne and olefin versus >27.0 kcal mol−1 for the pathway of forming Markovnikov-type products. The computational results are consistent with the experimental observations of Corma and co-workers. Furthermore, comparing the different metal catalysts of silver and copper for the activity and regioselectivity of the hydrothiolation, the results indicate that the silver catalyst has a relatively high catalytic activity, but the regioselectivity is not satisfactory compared to the gold catalyst and the regioselectivity of copper catalysts is the worst.
1. Introduction
Hydrothiolation of carbon–carbon unsaturated bonds is a simple and atom economical method of forming C–S bonds. Compounds containing S–C bonds have significant biological activity. Further, the sulfur–carbon bonds in pharmaceuticals, materials, synthetic reagents, and other natural products have a large number of applications.1–5 Vinyl sulfides have already been widely exploited in Diels–Alder,6 thio-Claisen,7 Michael acceptors8 and olefin metathesis.9 To our knowledge, many transition metals such as Ni, Pd, Pt, Co,Rh, Ir, Ru, Mo, Cu, Au, Zr, In,10–21 Ln (La, Sm, Y, Nd, Lu) and An (Th, U)22 are successfully applied to catalyze hydrothiolation for selective products. Rh, Ir and Au catalysts often favor anti-Markovnikov products, while Pt, Pd, Ni and Zr catalysts favor the Markovnikov vinyl sulfides. So far, some theoretical studies of hydrothiolation have been reported. However, the stereoselectivity of hydrothiolation still faces many challenges.
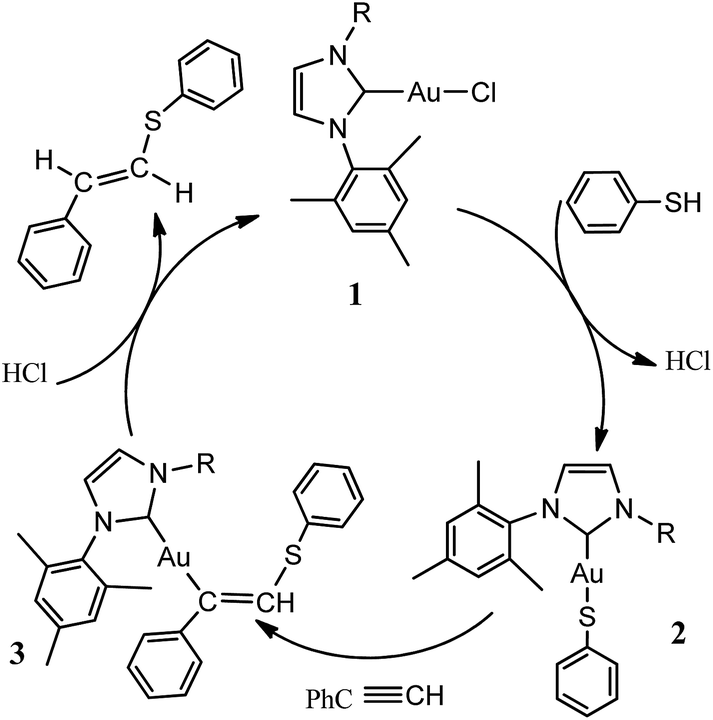
Recently, Krause and Morita reported23 the gold-catalyzed addition reaction of thiols and conjugated olefins to form C–S bond. Corma group24 studied N-heterocyclic carbene gold-catalyzed hydrothiolation of alkynes and electron-deficient olefin into anti-Markovnikov-type products in excellent yields in toluene solution at 40 °C (Scheme 1). According to the experimental results, the possible mechanism was postulated to explain the gold-catalyzed hydrothiolation of phenylacetylene with thiophenol as shown in Scheme 2. To our knowledge, metal-catalyzed hydrothiolation has not only been the subject of numerous theoretical studies but has also been practically applied to organic chemistry,25 there are no detailed theoretical studies for the gold-catalyzed hydrothiolation reported by the Corma et al.24 In this paper, in order to more in-depth discussion and interpretation of experimental results, we present a detailed DFT computational investigation of the mechanism and regioselectivity of the gold-catalyzed hydrothiolation to result in anti-Markovnikov-type or Markovnikov-type products on the basis of the experimental reported by Corma et al.24 In addition, DFT calculation results of gold-catalyzed hydrothiolation and Ag(I), Cu(I)-catalyzed hydrothiolation were compared, which will help the design and screening of selective catalyst.
 |
| | Scheme 1 Hydrothiolation of alkynes or olefins with thiophenol catalyzed by gold complexes. | |
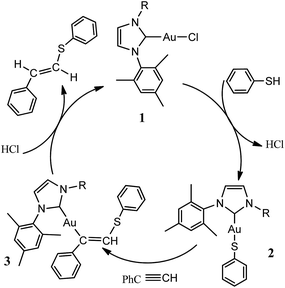 |
| | Scheme 2 Possible mechanisms envisioned for the gold-catalyzed phenylacetylene hydrothiolation with phenthiol. | |
2. Computational methods
The theoretical calculations reported here were carried out using the Gaussian 09 program suites.26 The geometries of all the stationary points were fully optimized by using density functional theory (DFT)27 based on the B3LYP28 method. This DFT method has been successfully applied in the mechanistic studies of gold-catalyzed reactions.29 In the DFT calculations, the Stuttgart–Dresden effective core potential (SDD)30 was employed with accurately account for relativistic effects to describe Au, Ag and Cu atoms. The 6-31G (d,p) basis set was utilized for S, Cl, N, C and H atoms. Vibrational analysis was performed at the same level to determine the character of the stationary points as either minima (the number of imaginary frequencies (NIMAG = 0) or transition states (NIMAG = 1)). The relative energies were thus corrected for the zero-point energies. In several significant cases, intrinsic reaction coordinate (IRC) calculations31 were performed to confirm that the transition states correctly connect the relevant reactants and products. To consider the effect of the solvent on the reactions of interest, the structures optimized was employed by B3LYP/6-311++G(d,p) (SDD for Au, Ag and Cu) single-point calculations with solvent correction (toluene, ε = 2.374) using the integral equation IEF-PCM protocol. The SMD solvation model32 was used, which have been reported by Truhlar and coworkers to be more accurate in calculating the solvation free energy.
3. Results and discussion
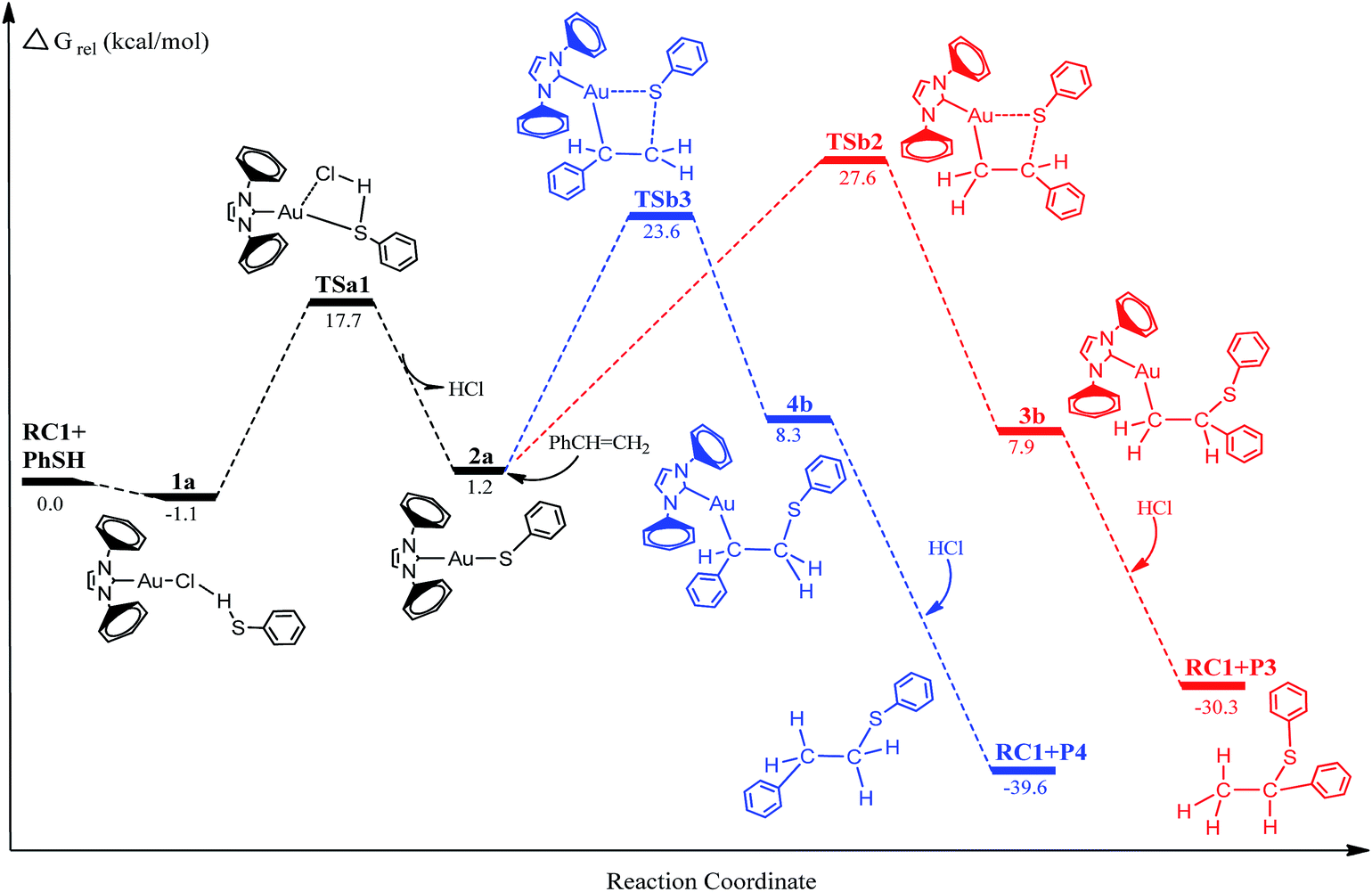
Energy profiles for the Au(I)-catalyzed hydrothiolation are shown in Fig. 1 and 2. The optimized geometries for the reactants (RC and PhSH), intermediates, transition states, and products of the reaction pathways are depicted schematically in Fig. 3 along with selected key geometry parameters (e.g., bond lengths). The relative energies and free energies of the reaction systems in solution phases are shown in Table 1. Unless otherwise noted, free energies discussed in subsequent sections refer to the values in toluene solvent. In order to keep the computational cost low, 1,3-biphenyl-imidazol-2-ylidene was selected as a model of the Au(I) catalyst ligands. The detailed structural parameters and energies for the structures determined here are collected in the ESI.†
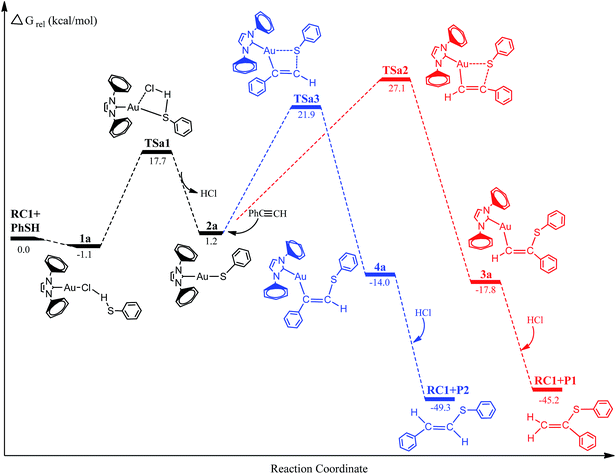 |
| | Fig. 1 Energy profiles for the hydrothiolation of phenylacetylene with thiophenol catalyzed by gold complexes. | |
 |
| | Fig. 2 Energy profiles for the hydrothiolation of styrene with thiophenol catalyzed by gold complexes. | |
 |
| | Fig. 3 Optimized structures for selected intermediates and transition states shown in Fig. 1 and 2 with selected structural parameters (hydrogen atoms are omitted for clarity and bond lengths are presented in Å). | |
Table 1 Relative energies and activation free energies in the gas phase and in solution of the key structures
| Species |
ΔErelgas |
ΔErelsol |
ΔGrelgas |
ΔErelsol |
| TSa2 |
26.2 |
25.9 |
27.6 |
27.1 |
| TSa3 |
22.0 |
21.5 |
21.6 |
21.9 |
| TSb2 |
28.9 |
28.7 |
27.7 |
27.6 |
| TSb3 |
24.4 |
24.2 |
23.3 |
23.6 |
| TSc2 |
24.7 |
24.3 |
24.5 |
24.6 |
| TSc3 |
20.2 |
20.6 |
21.0 |
21.3 |
| TSd2 |
23.7 |
23.8 |
23.1 |
22.9 |
| TSd3 |
23.3 |
23.5 |
22.2 |
22.4 |
3.1 Gold-catalyzed hydrothiolation of thiophenol with phenylacetylene
Free energy profile for gold-catalyzed hydrothiolation of thiophenol with phenylacetylene is depicted in Fig. 1. The structures of the various critical points located on the potential surface along with the most relevant geometry parameters are shown in Fig. 3. As can be seen from Fig. 1, two different reaction pathways for hydrothiolation were found to generate Markovnikov-type and anti-Markovnikov-type products, respectively. The first step involves a precursor complex 1a, where the hydrogen atom of thiophenol interacts with the chlorine atom of gold(I) catalyst (RC1). The preliminary complex 1a is formed without any barrier and is 1.1 kcal mol−1 lower in energy than the reactants [(RC1 + PhSH)]. In the structure 1a, the newly formed Cl–H bond length is 2.554 Å, Au–Cl distance is 2.338 Å. Meanwhile, the S–H bond has elongated 0.007 Å and S–H bond has been slightly activated. Subsequently, the intermediate 2a is formed and released a HCl molecule through the sulfur-transfer transition state TSa1 (TSa1 has only one imaginary frequency of 173.4 i cm−1 and IRC calculations confirmed that this TS connects the corresponding precursor complex and the intermediate). The corresponding imaginary frequency vibration mode is mainly reflected in the Cl–H bond in TS1a. From Fig. 3, the Au–Cl and S–H bonds in TS1a have become almost broken, bond lengths are 2.790 and 1.393 Å, and the new Au–S bond has been formed (2.529 Å). Examination of Fig. 1 and Table 1 show that activation free energy for this step is calculated to be 17.7 kcal mol−1 for TSa1 and the energy for the 2a intermediate is 1.2 kcal mol−1 with respect to the reactants. In 2a, the new Au–S bond has completely formed and is now 2.337 Å. These changes in the structure of the transition state TSa1 contribute to the formation of Au–S bond in 2a.
Inspection of Fig. 1 shows that the selective attack of a C1 atom and C2 atom of phenylacetylene on the Au–S bond of the 2a leads to the intermediates 3a and 4a through two different four-membered ring transition states TSa2 and TSa3, respectively. Vibrational analysis showed that TSa2 and TSa3 have only one imaginary frequency of 132.0 and 209.7 i cm−1, respectively. In the transition states TSa2 and TSa3, the new Au–C1, S–C2 and Au–C2, S–C1 bond lengths are 2.116, 2.966 Å and 2.148, 2.708 Å, respectively. As the reaction goes from TSa2 and TSa3 to 3a and 4a, the Au–S bonds have become completely broken from 2.905 and 3.110 Å to >3.450 Å, respectively. Table 1 and Fig. 1 show that the activation free energies for the TSa2 and TSa3 were 27.1 and 21.9 kcal mol−1 and the formation of 3a and 4a were also the exothermic process (the energies for the 3a and 4a were 19.0 and 15.2 kcal mol−1 with respect to 2a). This step also is the rate-determining step of the whole catalytic cycle process. The lower barriers were found for TSa3, and the energy barrier of TSa2 was higher than that of TSa3 by 5.2 kcal mol−1, with good stereoselectivity. The last step for the intermediates 3a and 4a are attacked by a molecular of HCl to results in the formation of the final product (Markovnikov-type product (P1) and anti-Markovnikov-type product (P2)) and regeneration of the catalyst (RC1). The steps take place without any barriers and are 27.7 and 35.3 kcal mol−1 lower than 3a and 4a, respectively. The whole catalytic processes are exothermic by 45.2 and 49.3 kcal mol−1 lower than reactants.
According to experimental results, gold(I)-catalyzed hydrothiolation of thiophenol with phenylacetylene in toluene as a solvent, 0.1 mol% of catalyst at a temperature at 40 °C reaction conditions, can be obtained 90–97% of the anti-Markov product, which has good anti-Markov regioselectivity. The results of theoretical calculations show that the reaction channel of anti-Markov product generated has a relatively low activation free energy, and the computational results also show good regioselectivity. The theoretical results are in good agreement with the experimental observations of Corma et al.24
3.2 Gold-catalyzed hydrothiolation of thiophenol with styrene
Experimental studies show that gold(I)-catalyzed hydrothiolation of electron-deficient olefin would take place with good regioselectivity and high yield. Following the originally proposed mechanism and the reaction system, the detailed mechanism for gold-catalyzed hydrothiolation of thiophenol with styrene is displayed in Fig. 2. The structures of the critical points to situate on the potential surface along with the key geometry parameters are presented in Fig. 3. Similar to the pathways of Fig. 1, the gold complex intermediate 2a also should be formed. Fig. 2 shows that the subsequent step involves the double bonds of styrene insertion of Au–S bond and gives the new, stable intermediate structures 3b and 4b through the four-membered ring transition structures TSb2 and TSb3, respectively. In Fig. 3, the Au–C bond has been formed (TSb2 in Au–C1 is 2.160 Å; TSb3 in Au–C2 is 2.208 Å) in the transition state structure. The C1–C2 bond of olefin has also been shortened, and gradually reflected some single bond character (the C1–C2 bonds are now 1.426 Å in TSb2 and 1.416 Å in TSb3). The structures of TSb2 and TSb3 indicate that the sp2 → sp3 rehybridization migration for C1 and C2 atoms has been carried out. As the reaction goes from the transition states TSb2 and TSb3 to the intermediate 3b and 4b, it is evident that the S–C bond has completely connected and the Au–S bond becomes completely broken. The C1–C2 bond has lost a small amount of its double-bond feature, which is now 1.546 and 1.539 Å in 3b and 4b, respectively. In Table 1 and Fig. 2 the activation free energy is calculated to be 27.6 kcal mol−1 for TSb2 and 23.6 kcal mol−1 for TSb3. It is important to point out that this step also is the rate-determining step for the whole reaction. The higher barriers found for TSb2 and TSb3 in comparison to those for TSa2 and TSa3 can be mainly attributed to electronegativity of alkyne above olefin. The subsequent the intermediates 3b and 4b undergo HCl attack and result in the formation of the final products (P3 and P4) and regeneration of the catalyst (RC1). The whole catalytic process is exothermic by 30.3 and 39.6 kcal mol−1 lower than the reactants, respectively. The reaction system also tends to generate anti-Markov product, consistent with the experimental findings.
3.3 Comparative reactivity of Au(I), Ag(I), Cu(I)-catalyzed hydrothiolation of thiophenol with phenylacetylene
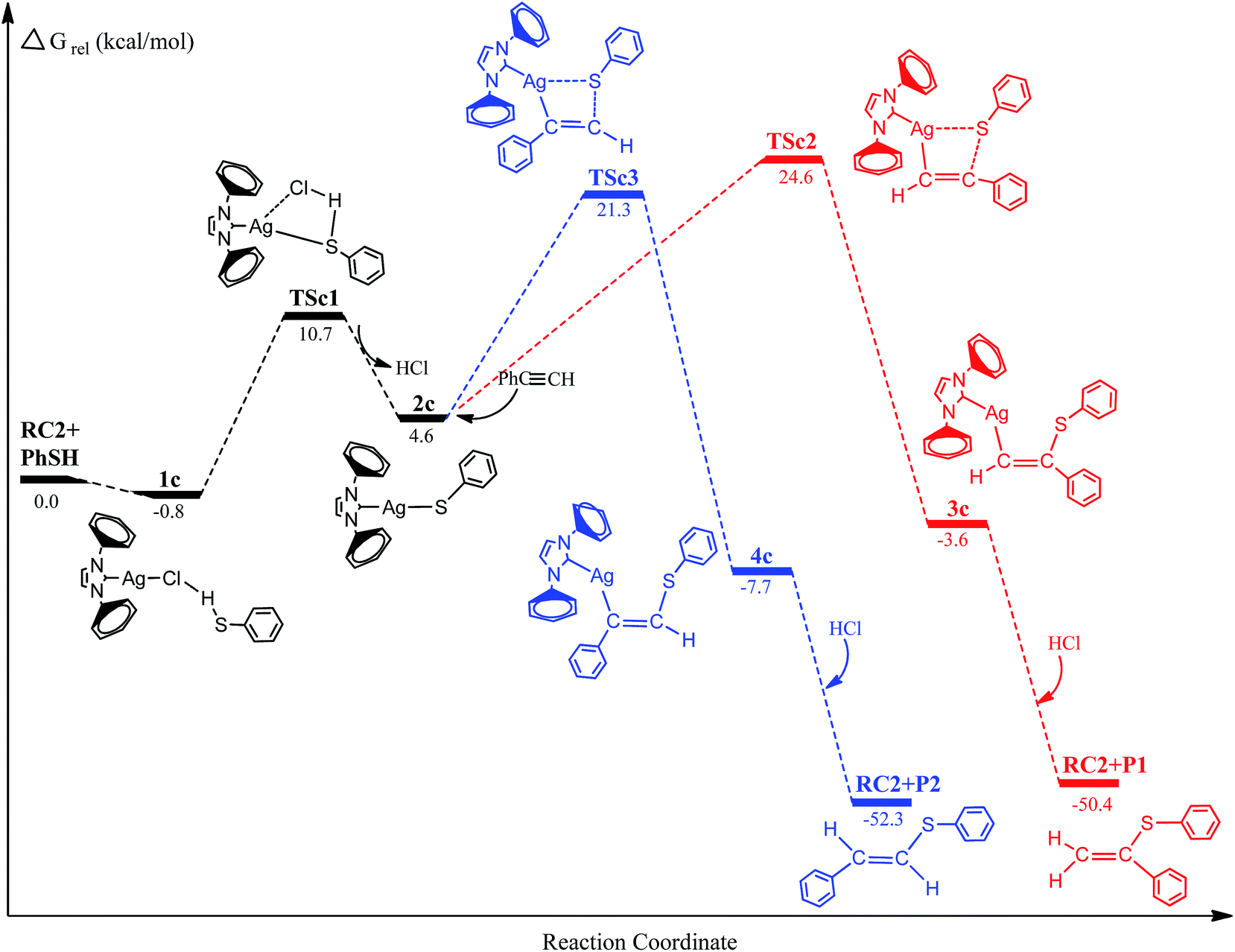
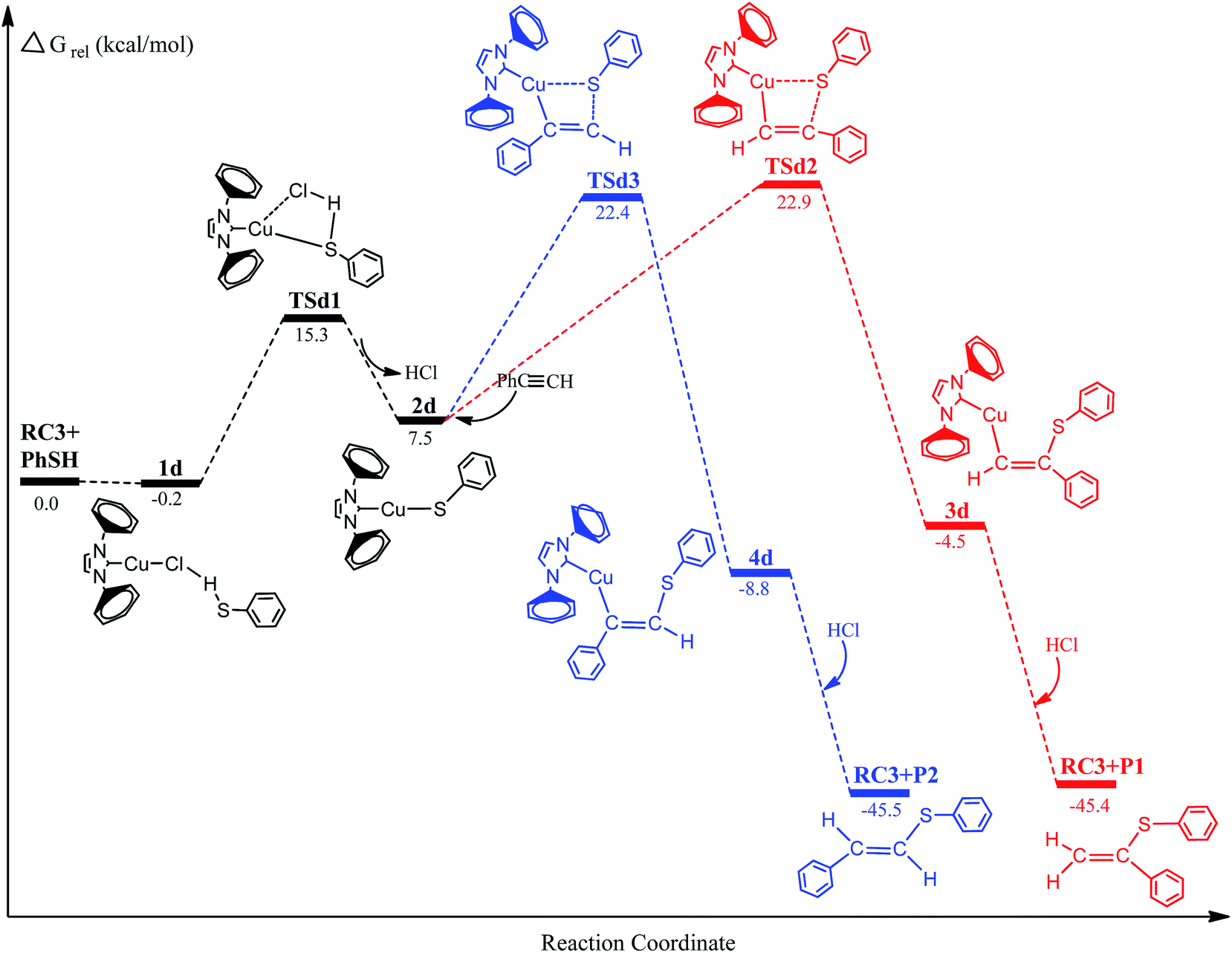
To examine further the effect of the reaction mechanism and the reaction activity about different metal-activator, we have chosen the reaction systems of (IPr)MCl (M = Cu and Ag) to catalyze hydrothiolation of thiophenol with phenylacetylene as described in Fig. 4 and 5. The similar reaction pathways have been determined for these reaction systems. The second step of the reaction systems is still the rate-determining step for the catalytic cycle process. Inspection of Fig. 4, 5 and Table 1 shows that the activation free energies for Ag (TSc2: 24.6 kcal mol−1; TSc3: 21.3 kcal mol−1) and Cu (TSd2: 22.9 kcal mol−1; TSd3: 22.4 kcal mol−1) are lower than Au (TSa2: 27.1 kcal mol−1; TSa3: 21.9 kcal mol−1) catalyst. Comparison of activation free energies for the Au(I), Ag(I) and Cu(I)-catalyzed hydrothiolation of thiophenol with phenylacetylene is shown in Fig. 6. The results have shown that the Ag catalyst has the highest catalytic activity, but the regioselectivity of the products is not very satisfactory. In addition, Cu catalysis has a relatively high activity, but the activation free energies of two reaction channels are almost no difference, there is no regioselectivity. The computed energetics suggest that the reaction steps of will Ag(I)-catalyzed hydrothiolation much more easily occur than that of Au(I) and Cu(I)-catalyzed hydrothiolation, while the regioselectivity of Au(I)-catalyzed hydrothiolation is best. Because Lewis acidity compared with three metal species for many transformations is Ag > Cu > Au.33 The higher the acidity is the more favorable for the hydrothiolation process. These results predict that (IPr)AgCl could be one of the highly chemical reactivity reagents, and (IPr)AuCl is the efficient catalyst of anti-Markov product for the hydrothiolation.
 |
| | Fig. 4 Energy profiles for the hydrothiolation of phenylacetylene with thiophenol catalyzed by silver complexes. | |
 |
| | Fig. 5 Energy profiles for the hydrothiolation of phenylacetylene with thiophenol catalyzed by copper complexes. | |
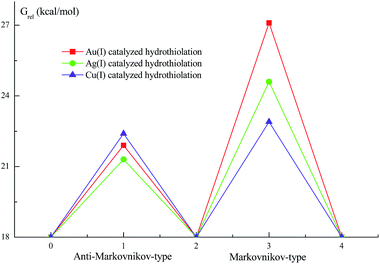 |
| | Fig. 6 Comparison of activation free energies for the Au(I), Ag(I) and Cu(I)-catalyzed hydrothiolation of thiophenol with phenylacetylene. | |
4. Conclusion
In summary, this work has provided the first theoretical investigation at the DFT (B3LYP) level for the gold(I)-catalyzed hydrothiolation of thiophenol with phenylacetylene and styrene. The calculations suggest that the first step of the catalytic cycle is the proton-transfer process from thiophenol onto the chlorine atom to form the gold–sulfur intermediate through the four-membered ring transition state. Next, migratory insertion of the alkyne and olefin into the Au–S bond is the rate-determining step of the whole cycle. The results shown that the gold-catalyzed hydrothiolation of thiophenol with phenylacetylene and styrene to generate anti-Markov product has the lower free energy of activation (21.9 and 23.6 kcal mol−1, respectively), and the anti-Markov product is widely favored, while the free energy for the Markov product are relatively higher (>27.0 kcal mol−1). The gold-catalyzed hydrothiolation has the good regioselectivity to generate the anti-Markov product. The calculated results are consistent with the experimental observations of Corma group for the gold-catalyzed hydrothiolation of thiophenol with phenylacetylene and styrene. According to the result of experiment, the paper calculated and compared the activation free energies of the (IPr)MCl (M = Cu, Ag or Au) catalyzed hydrothiolation. The trend is similar to the case of the gold catalytic process. Meanwhile, the results of compared with (IPr)MCl (M = Cu, Ag or Au) predict that Ag(I) catalyst could be one of the highly chemical reactivity hydrothiolation reagents, while the regioselectivity of the products is not very good, which has comprehensive ramifications for experimental design and synthesize.
Acknowledgements
This work was supported by Longyuan young creative talents to support projects, Gansu Province (2014-98), and New Chemical Materials Research and Innovation team project. We are grateful to the Gansu Province Supercomputer Center for essential support. We are grateful to the reviewers for their invaluable suggestions.
Notes and references
-
(a) L. SanMiguel and A. J. Matzger, Macromolecules, 2007, 40, 9233 CrossRef CAS;
(b) J. Roncali, Chem. Rev., 1992, 92, 711 CrossRef CAS.
-
(a) M. A. Cortez and S. M. Grayson, Macromolecules, 2010, 43, 4081 CrossRef CAS;
(b) D. Konkolewicz, A. GrayWeale and S. B. Perrier, J. Am. Chem. Soc., 2009, 131, 18075 CrossRef CAS PubMed;
(c) C. J. Kloxin, T. F. Scott and C. N. Bowman, Macromolecules, 2009, 42, 2551 CrossRef CAS PubMed;
(d) E. Ahmed, F. S. Kim, H. Xin and S. A. Jenekhe, Macromolecules, 2009, 42, 8615 CrossRef CAS;
(e) B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2008, 42, 211 CrossRef PubMed;
(f) A. Valdebenito and M. V. Encinas, Polymer, 2005, 46, 10658 CrossRef CAS PubMed.
-
(a) Z. Ma, L. Rao and U. Bierbach, J. Med. Chem., 2009, 52, 3424 CrossRef CAS PubMed;
(b) A. Szilagyi, F. Fenyvesi, O. Majercsik, I. F. Pelyvas, I. Bacskay, P. Feher, J. Varadi, M. Vecsernyes and P. Herczegh, J. Med. Chem., 2006, 49, 5626 CrossRef CAS PubMed;
(c) P. Johannesson, G. Lindeberg, A. Johansson, G. V. Nikiforovich, A. Gogoll, B. Synnergren, M. Le Greves, F. Nyberg, A. Karlen and A. Hallberg, J. Med. Chem., 2002, 45, 1767 CrossRef CAS PubMed;
(d) L. Rocheblave, F. Bihel, C. De Michelis, G. Priem, J. Courcambeck, B. Bonnet, J. C. Chermann and J. L. Kraus, J. Med. Chem., 2002, 45, 3321 CrossRef CAS PubMed.
-
(a) D. Meng, W. Chen and W. Zhao, J. Nat. Prod., 2007, 70, 824 CrossRef CAS PubMed;
(b) Y. Zhang, S. Liu, Y. Che and X. Liu, J. Nat. Prod., 2007, 70, 1522 CrossRef CAS PubMed;
(c) K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis, Wiley-VHC, New York, 1996 Search PubMed.
-
(a) C. A. Dvorak, W. D. Schmitz, D. J. Poon, D. C. Pryde, J. P. Lawson, R. A. Amos and A. I. Meyers, Angew. Chem., Int. Ed., 2000, 39, 1664 CrossRef CAS;
(b) M. Ceruti, G. Balliano, F. Rocco, P. Milla, S. Arpicco, L. Cattel and F. Viola, Lipids, 2001, 36, 629 CrossRef CAS PubMed;
(c) P. Johannesson, G. Lindeberg, A. Johansson, G. V. Nikiforovich, A. Gogoll, B. Synnergren, M. LeGreves, F. Nyberg, A. Karlen and A. Hallberg, J. Med. Chem., 2002, 45, 1767 CrossRef CAS PubMed.
-
(a) S. S. P. Chou and S. J. Wey, J. Org. Chem., 1990, 55, 1270 CrossRef CAS;
(b) S. R. Surasani and R. K. Peddinti, Tetrahedron Lett., 2011, 52, 4615 CrossRef CAS PubMed.
-
(a) N. W. Boaz and K. M. Fox, J. Org. Chem., 1993, 58, 3042 CrossRef CAS;
(b) K. C. Majumdar, U. K. Kundu and S. K. Ghosh, Org. Lett., 2002, 4, 2629 CrossRef CAS PubMed;
(c) R. Fernandez de la Pradilla, M. Tortosa and A. Viso, Top. Curr. Chem., 2007, 275, 103 CrossRef CAS;
(d) Z. Liu, S. J. Mehta, K. S. Lee, B. Grossman, H. Qu, X. Gu, G. S. Nichol and L. J. Hruby, J. Org. Chem., 2012, 77, 1289 CrossRef CAS PubMed.
- H. Mizuno, K. Domon, K. Masuya, K. Tanino and I. Kuwajima, J. Org. Chem., 1999, 64, 2648 CrossRef CAS PubMed.
-
(a) Z. Liu and J. D. Rainier, Org. Lett., 2005, 7, 131 CrossRef CAS PubMed;
(b) M. L. Macnaughtan, J. B. Gary, D. L. Gerlach, M. J. A. Johnson and J. W. Kampf, Organometallics, 2009, 28, 2880 CrossRef CAS.
- U. Koelle, C. Rietmann, J. Tjoe, T. Wagner and U. Englert, Organometallics, 2005, 14, 703 CrossRef.
-
(a) S. Burling, L. D. Field, B. A. Messerle, K. Q. Vuong and P. Turner, Dalton Trans., 2003, 4181 RSC;
(b) C. Cao, L. R. Fraser and J. A. Love, J. Am. Chem. Soc., 2005, 127, 17614 CrossRef CAS PubMed;
(c) Y. Misumi, H. Seino and Y. J. Mizobe, Organomet. Chem., 2006, 691, 3157 CrossRef CAS PubMed;
(d) L. R. Fraser, J. Bird, Q. Wu, C. Cao, B. O. Patrick and J. A. Love, Organometallics, 2007, 26, 5602 CrossRef CAS;
(e) S. Shoai, P. Bichler, B. Kang, H. Buckley and J. A. Love, Organometallics, 2007, 26, 5778 CrossRef CAS;
(f) J. Yang, A. Sabarre, L. R. Fraser, B. O. Patrick and J. A. Love, J. Org. Chem., 2009, 74, 182 CrossRef CAS PubMed;
(g) Y. Yang and R. M. Rioux, Chem. Commun., 2011, 47, 6557 RSC;
(h) J. Liu, J. W. Y. Lam, C. K. W. Jin, J. C. Y. Ng, J. Shi, H. Su, K. F. Yeung, Y. Hong, M. Faisal, Y. Yu, K. S. Wong and B. Z. Tang, Macromolecules, 2011, 44, 68 CrossRef CAS;
(i) H. Zhao, J. Peng and M. Cai, Catal. Lett., 2012, 142, 138 CrossRef CAS.
- L. D. Field, B. A. Messerle, K. Q. Vuong and P. Turner, Dalton Trans., 2009, 3599 RSC.
-
(a) L. B. Han, C. Zhang, H. Yazawa and S. Shimada, J. Am. Chem. Soc., 2004, 126, 5080 CrossRef CAS PubMed;
(b) V. P. Ananikov, L. V. Orlov and I. P. Beletskaya, Organometallics, 2006, 25, 1970 CrossRef CAS;
(c) D. A. Malyshev, N. M. Scott, N. Marion, E. D. Stevens, V. P. Ananikov, I. P. Beletskaya and S. P. Nolan, Organometallics, 2006, 25, 4462 CrossRef CAS;
(d) V. P. Ananikov, K. A. Gayduck, N. V. Orlov, I. P. Beletskaya, V. N. Khrustalev and M. Y. Antipin, Chem.–Eur. J., 2010, 16, 2063 CrossRef CAS PubMed.
-
(a) H. Kuniyasu, A. Ogawa, K. I. Sato, I. Ryu, N. Kambe and N. Sonoda, J. Am. Chem. Soc., 1992, 114, 5902 CrossRef CAS;
(b) J. E. Backvall, J. Org. Chem., 1994, 59, 5850 CrossRef;
(c) A. Ogawa, T. Ikeda, K. Kimura and J. Hirao, J. Am. Chem. Soc., 1999, 121, 5108 CrossRef CAS;
(d) A. Kondoh, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9, 1383 CrossRef CAS PubMed;
(e) V. P. Ananikov, N. V. Orlov, I. P. Beletskaya, V. N. Khrustalev, M. Y. Antipin and T. V. Timofeeva, J. Am. Chem. Soc., 2007, 129, 7252 CrossRef CAS PubMed;
(f) T. Mitamura, M. Daitou, A. Nomoto and A. Ogawa, Bull. Chem. Soc. Jpn., 2011, 84, 413 CrossRef CAS.
-
(a) A. Ogawa, J. I. Kawakami, M. Mihara, T. Ikeda, N. Sonoda and T. Irao, J. Am. Chem. Soc., 1997, 119, 12380 CrossRef CAS;
(b) V. P. Ananikov and I. P. Beletskaya, Pure Appl. Chem., 2007, 79, 1041 Search PubMed.
- J. V. McDonald, J. L. Corbin and W. E. Newton, Inorg. Chem., 1976, 15, 2056 CrossRef CAS.
- Y. Higuchi, S. Atobe, M. Tanaka, I. Kamiya, T. Yamamoto, A. Nomoto, M. Sonoda and A. Ogawa, Organometallics, 2011, 30, 4539 CrossRef CAS.
- S. A. Delp, C. Munro-leighton, L. A. Goj, M. A. Ramírez, T. B. Gunnoe, J. L. Petersen and P. D. Boyle, Inorg. Chem., 2007, 46, 2365 CrossRef CAS PubMed.
- A. Corma, C. Gonzalez-Arellano, M. Iglesias and F. Sanchez, Appl. Catal., A, 2010, 375, 49 CrossRef CAS PubMed.
- C. J. Weiss and T. J. Marks, J. Am. Chem. Soc., 2010, 132, 1053 Search PubMed.
- J. S. Yadav, B. V. S. Reddy, A. Raju, K. Ravindar and G. Baishya, Chem. Lett., 2007, 36, 1474 CrossRef CAS.
-
(a) C. J. Weiss, S. D. Wobser and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 2062 CrossRef CAS PubMed;
(b) C. J. Weiss, S. D. Wobser and T. J. Marks, Organometallics, 2010, 29, 6308 CrossRef CAS.
- N. Morita and N. Krause, Angew. Chem., Int. Ed., 2006, 45, 1897 CrossRef CAS PubMed.
- A. Corma, C. González-Arellano, M. Iglesias and F. Sánchez, Appl. Catal., A, 2010, 375, 49 CrossRef CAS PubMed.
-
(a) X. H. Zhang, Z. Y. Geng, Y. C. Wang, X. F. Hou and D. M. Wang, J. Mol. Catal. A: Chem., 2012, 363–364, 31 CrossRef CAS PubMed;
(b) X. H. Zhang, Z. Y. Geng, K. T. Wang and S. S. Li, J. Mol. Model., 2014, 20, 2409 CrossRef PubMed;
(c) A. D. Giuseppe, R. Castarlenas, J. Perez-Torrente, M. Crucianelli, V. Polo, R. Sancho, F. J. Lahoz and L. A. Oro, J. Am. Chem. Soc., 2012, 134, 8171 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, revision A.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. G. Parr and W. Yang, Density-functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
-
(a) E. Soriano and J. Marco-Contelles, Acc. Chem. Res., 2009, 42, 1026 CrossRef CAS PubMed;
(b) R. Fang and L. Z. Yang, Organometallics, 2012, 31, 3043 CrossRef CAS.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
-
(a) S. Ma and J. Zhang, Angew. Chem., Int. Ed., 2003, 42, 184 Search PubMed;
(b) S. Ma, L. Lu and J. Zhang, J. Am. Chem. Soc., 2004, 126, 9645 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables giving Cartesian coordinates for the calculated stationary structures obtained from the DFT calculations. See DOI: 10.1039/c5ra01883h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.