
Fire and mechanical properties of a novel free-radically cured phenolic resin based on a methacrylate-functional novolac and of its blends with an unsaturated polyester resin
Baljinder K. Kandola*,
Latha Krishnan,
Dario Deli†
,
Piyanuch Luangtriratana and
John R. Ebdon
Fire Materials Group, Institute for Materials Research and Innovation (IMRI), University of Bolton, Deane Road, Bolton, BL3 5AB, UK. E-mail: b.kandola@bolton.ac.uk
First published on 2nd April 2015
Abstract
A novel phenolic novolac resin bearing methacrylate functional groups has been synthesized by reaction of the novolac with methacryloyl chloride. This resin has been mixed with styrene and cured (crosslinked) free-radically under the relatively low temperature conditions used to cure unsaturated polyester/styrene mixtures, i.e. there is no need to employ the high temperatures and pressures that are required to cure conventional phenolic resins. Homogeneous cured blends of the methacrylated novolac with unsaturated polyester and styrene have been prepared also. The cured methacrylated novolac, and its blends with unsaturated polyester, are rigid materials with good mechanical strength, and have glass transition temperatures, thermal stabilities and flame retardancies superior to those of cured unsaturated polyester alone.
Introduction
Phenol–formaldehyde resins, known simply as phenolic resins (PH) are considered as inherently fire retardant resins and are used in applications where fire performance is an important criterion. Since their first development in 1907 (ref. 1) further modifications to the chemistry and molecular structure of these resins have resulted in an increase in their applications ranging from commodity and construction materials, such as thermal insulation materials, moulding compounds, foundry binders, wood adhesives and coatings, to high technology aerospace materials, such as fibre-reinforced composites.2 In the composites area, they cannot however compete with epoxies, polyimides, etc. for primary structural components, and are mainly used for non load-bearing structures such as thin laminates or sandwich panels. This is due to their brittle nature leading to low poor mechanical properties. PH resins cross-link by polycondensation with the elimination of water and formaldehyde.3–6 During curing of the resins at elevated temperatures, these vapours cause some voids and surface defects,5 which reduce the strength of composites in which these resins are used as matrices. This problem is particularly acute in conventional thick composites, cured by conventional thermal processes, hence limiting their usage in structural composites. This problem however, can be addressed by modifying the resin and/or cure chemistry so that they can cross-link without production of volatiles.While different types of phenolic resins are available, classical resole and novolac types dominate the resin market. Phenol–formaldehyde (P/F) molar ratio and catalyst type (acid/alkali) used to synthesize a resin determines the resole/novolac resin type.6 Resins produced with P/F < 1 and alkaline catalyst are referred to as resoles. Resoles contain reactive methylol or dimethylene–ether linkages and can be cured by applying heat. Novolacs on the other hand are synthesized using P/F > 1 under acidic conditions and consist of phenol rings connected solely by methylene bridges. Novolac resins are thermally cured by addition of a methylene crosslinker such as hexamethylenetetramine.7,8 Resoles, being liquids, are easy to handle, hence are preferred for fibre-reinforced composites. Novolacs on the other hand are stable, thermoplastic, solid resins, and are preferred for moulding materials or as a component of other systems such as of epoxies. However, both resin types, on curing, produce highly cross-linked thermally stable networks, which on exposure to high heat or fire char so producing relatively low levels of combustible volatiles; hence their flammabilities are lower than those of epoxies and unsaturated polyester resins.
In our on-going research at Bolton we are exploring blending different phenolic resins with unsaturated polyester resin to reduce the flammability of the latter while maintaining its physical and mechanical properties for potential use in marine composites.4,9–12 We have adopted this approach to improving the fire retardance and also the mechanical properties of cured UP resins, rather than use perhaps more obvious strategies, such as adding a non-combustible inorganic filler, in order to preserve the low viscosity of the uncured resin, which is necessary if composite laminates are to be prepared from these resins by pressurized resin infusion, a method commonly used for the preparation of composite laminate components in the marine industry. While resin blending is well-established for thermosetting resins with compatible cure chemistries, such as epoxy and phenolic resins,13 it is challenging for incompatible systems such as UP and phenolics, owing to the inherent physical incompatibility of relatively hydrophobic UP resins with relatively hydrophilic formaldehyde-based resins, and also to the fact that the former cure by a low-temperature (typically 40–80 °C) free-radical chain reaction whereas the latter cure via a high temperature (ca. 150 °C) polycondensation, often with an acid catalyst.9,10,14,15 We have done extensive work on resole phenolic–UP blends4,9–12 and overcame these problems by compatibilizing resoles using different approaches: (i) the use of a common solvent (ethanol), (ii) using an external compatibilizer (epoxy functionalised phenolic resin) and (iii) chemical functionalization of at least one of the components of the blend (use of an allyl functionalized phenolic resin). These compatibilized, co-cured, resin blends are significantly more flame retardant than unmodified UPs, burn with lower evolution of heat and the formation of more char, have physical and mechanical properties that are superior to those of unmodified UPs, and can satisfactorily be used to make glass-reinforced composite panel.4,10–12 Of the modified resoles, that bearing allyl groups (a commercial Methylon resin) was found to be the most satisfactory in forming homogeneous blends with UP owing to the chemical incorporation of at least some of the allyl groups into the free-radically crosslinked network structure. However, allyl-functional resoles are designed primarily to be used as high temperature self-curing surface coating materials and, in our formulations, a high temperature post-curing process is still required to give dimensionally stable materials (i.e. to condense residual methylol groups in the resole and to complete polymerization of the allyl groups).4,10
In this paper we report the chemical modification of a PH novolac, i.e. a phenolic resin with no free methylol groups, with methacryloyl chloride to introduce methacrylate groups. These groups too were expected to aid miscibility of the PH resin with UP/styrene but to be more reactive in free-radical reactions than allyl groups and so permit the PH novolac to be free-radically cured with styrene and in blends with UP/styrene under conditions similar to those used for UP/styrene alone, i.e. room temperature for 24 h and post curing at 80 °C for 6 h.
Surprisingly there seems to have been relatively little work before to modify phenolic resins so as to enable them to take part in free-radical polymerization reactions, although acrylate and methacrylate functional epoxy novolacs, made by ring-opening reactions of epoxy groups in the novolac with acrylic or methacrylic acid, have been known for 50 years16 and are used extensively today in photocurable surface coatings and inks.17 Also there are reports of phenolics modified so as to contain vinyl benzyl and maleimide groups, also capable of being free radically polymerized.18,19
Materials and method
Materials
Crystic 2-406 PA, Scott-Bader: a phthalic anhydride-based unsaturated polyester (UP) resin containing 0.015% by weight of Accelerator G (cobalt(II) octoate), 0.3% by weight of di-methyl aniline, and ca. 35% by weight of styrene.Catalyst M, Scott-Bader: a free-radical catalyst for UP curing consisting of methyl ethyl ketone peroxide dissolved in methyl ethyl ketone.
Accelerator G, Scott Bader: 1% by weight cobalt(II) octoate dissolved in styrene.
Durez 31459, Sumitomo Bakelite Europe N.V.: a phenolic novolac resin (Nov), MW = 2500.
Triethylamine (TEA), styrene and di-methyl aniline (DMA), TCI Europe.
Methacryloyl chloride (MC) and anhydrous tetrahydrofuran (THF), Aldrich.
Preparation of methacrylated novolac resin (M-Nov)
Novolac D31459 (Nov) (50 g, containing 0.4717 mol of reacted phenol) was dissolved in anhydrous THF (120 ml) in a 700 ml quick-fit round bottom flask purged with nitrogen and equipped with condenser, thermometer and mechanical stirrer. During dissolution, the temperature was slowly raised to 60 °C. When the dissolution was complete, TEA (57.3 g, 0.5660 mol) was added slowly to the Nov solution (so as not to avoid precipitation of the novolac). MC (59.1 g, 0.5660 mol) was dissolved in anhydrous THF (50 ml) and added dropwise to the reaction mixture. The reaction is exothermic and slow addition is required to keep the temperature no higher than 60 °C. After 24 h, the reaction mixture was cooled to RT. The mixture, which contained a white precipitate of TEA hydrochloride (TEA·HCl), was then filtered under vacuum using a Buchner funnel equipped with glass microfiber filter (0.7 μm porosity). The reaction scheme is depicted in Fig. 1. | ||
| Fig. 1 Reaction of Nov with methacryloyl chloride to give M-Nov. | ||
The recovered TEA·HCl was washed with THF (3 × 20 ml) and dried in a vacuum oven for 24 hours at 80 °C. The dried TEA·HCl was weighed in order to determine the yield and thus indirectly the extent of methacrylation. One ml of the organic solution containing the M-Nov was evaporated in an oven at 100 °C for 24 h in order to calculate the solid content and provide a true measure of yield. THF was then evaporated off under vacuum from the remaining solution of M-Nov at a temperature below 60 °C. Styrene (26 g) was added to the mixture, which was stirred at high shear and cooled to RT.
Yields: 76.6 g of TEA·HCl, corresponding to 98% conversion of novolac to methacrylated novolac (77.9 g being the theoretical amount of TEA·HCl produced for 100% methacrylation of phenolic groups in the novolac); 80.9 g of M-Nov, corresponding to 99% methacrylation. The characterization of the resin by IR and NMR spectroscopy both before and after methacrylation is discussed later.
Casting and curing of resins and resin blends
Samples of cured M-Nov and UP resins were prepared by mixing the resins with 2% w/w of Catalyst M with a mechanical stirrer in a 100 ml beaker. Prior to curing, 32% by weight of styrene was added with stirring to M-Nov to match the styrene content present in the UP resin and also to ensure that all blends of UP and M-Nov contained the same overall amounts of styrene (bearing in mind that Accelerator G is supplied as a solution in styrene) plus 0.015% by weight of Accelerator G and 0.3% by weight of di-methyl aniline. The mixtures were then poured into 5.5 cm diameter circular open aluminium moulds to a depth of 3 mm. The specimens were then allowed to cure at room temperature for 24 h and subsequently post-cured at 80 °C in an oven for 6 h. To obtain consistent, void-free, samples, it was found necessary, following the RT curing stage, to ramp the temperature up to 80 °C for the post-cure stage at no more than 3 °C min−1. The mechanism of curing in these systems is a free radical one, and is initiated at room temperature through a redox process in which the MEK peroxide, a complex mixture of monomeric, dimeric and oligomeric peroxides and hydroperoxides (Catalyst M), is catalytically decomposed by reaction with the cobalt(II) octoate (Accelerator G). One such decomposition, that of a hydroperoxide group, is illustrated schematically below:| ROOH + Co2+ → RO˙ + HO− + Co3+ |
Di methyl-aniline also catalyses the decomposition of MEK peroxide.
Resin blends were prepared by mixing 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 and 50:50% w/w UP/M-Nov for 10 min in a 100 ml beaker using a high-speed, overhead, electric stirrer fitted with a four-component blade (IKA RW16 at 900 rpm). Catalyst M (2% by weight w.r.t. resin blend) was then added to the resin mixtures and stirring continued for a further 10 min. The resulting mixtures were transferred to moulds and cured in the same way as for the pure resins. The appropriateness of the curing regimes outlined above was established by differential scanning calorimetric (DSC) studies of curing, the results of which are given in a later section.
:30 and 50:50% w/w UP/M-Nov for 10 min in a 100 ml beaker using a high-speed, overhead, electric stirrer fitted with a four-component blade (IKA RW16 at 900 rpm). Catalyst M (2% by weight w.r.t. resin blend) was then added to the resin mixtures and stirring continued for a further 10 min. The resulting mixtures were transferred to moulds and cured in the same way as for the pure resins. The appropriateness of the curing regimes outlined above was established by differential scanning calorimetric (DSC) studies of curing, the results of which are given in a later section.
Characterization
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra of solid samples were acquired using a Nicolet iS10 spectrometer equipped with a Smart iTR attachment employing a single bounce diamond crystal.Proton NMR spectra were recorded on solutions of Nov and M-Nov in acetone on a Bruker Avance 400 spectrometer at the University of Manchester. Solid-state C13 NMR spectra of cured samples of M-Nov, UP and UP/M-Nov blends were recorded on a Bruker Avance III HD spectrometer courtesy of the EPSRC-funded, University of Durham solid-state NMR service.
Differential scanning calorimetry (DSC) was used to monitor the curing of resin samples (2–10 mg) at a heating rate of 5 °C min−1 over the temperature range 30–350 °C using a TA Instruments Q2000 differential scanning calorimeter.
Dynamic mechanical thermal analysis (DMTA) was carried out on a TA Instruments Q800 DMA machine using a single cantilever clamp and multi-frequency-strain set-up (0.1% strain and 1 Hz frequency). The specimens were heated at 10 °C min−1 within the temperature range 30–350 °C. Values of tanδ and storage modulus were recorded.
Scanning electron microscopy (SEM) was performed on small samples of cured, cast resins previously fractured in simple bending experiments, and the fracture surfaces then gold coated using a Polaron Range SC7620 Sputter Coater with 60 s plasma exposure. The coated fracture surfaces were examined using an Hitachi S-3400N variable pressure scanning electron microscope.
Thermogravimetry-FTIR study
Thermogravimetric analyses (TGA) of all cured resins and their blends were performed on a TA Instruments SDT 2960 simultaneous DTA (differential thermal analysis)-TGA instrument from room temperature to 800 °C using 15 ± 1 mg samples heated at a constant rate of 10 °C min−1 in both nitrogen and air flowing at 100 ± 5 ml min−1. The experiments were performed in duplicate and showed good reproducibility. Averaged data are presented. During the experiments in nitrogen, the SDT 2960 was linked to a Nicolet Smart iS10-iTR FTIR spectrometer equipped with a gas cell for the analysis of gases evolved during decomposition.Flammability study
The limiting oxygen indices (LOI) for samples of cured resins and their blends were measured according to a standard method (BS 2782) using a LOI instrument (Fire Testing Technology Ltd) equipped with an oxygen analyzer. At least five specimens of dimensions 100 mm × 10 mm × ∼3 mm were tested for each resin sample.A cone calorimeter (Fire Testing Technology Ltd) was used to assess flammability parameters for cured resins according to ISO 5660 standard with the exception of sample size. Circular samples of cured resins measuring 5.5 cm dia. with a nominal thickness of 3 mm, were fire tested in the horizontal mode with an ignition source at an applied heat flux of 50 kW m−2. Before testing, the bottom surface and the edges of the samples were wrapped with aluminium foil to ensure that only the top surface would be directly exposed to the heat source. A minimum of three tests were performed for each formulation. Previously in our laboratories, a comparative study of the cone calorimetric behaviour of round and standard square (10 cm × 10 cm) samples was undertaken in order to understand the effect of geometry on flammability properties of polymeric materials.20 Circular specimens with a four-fold reduction in area gave similar results for the peak heat release rates (PHRR), total heat release (THR) and effective heat of combustion (EHC). Smoke, CO and CO2 production results were found to be different from those measured for the larger specimens since these parameters are independent of exposed specimen surface area. However, in this work, these data were used for comparison purposes only with respect to control specimens, hence there was no need for adjustments.
Results and discussion
ATR FT-IR analysis of Nov and M-Nov
ATR spectra of Nov and the M-Nov are shown in Fig. 2. Characteristic peaks in the spectrum of the unmodified Nov are evident at ca. 3310 cm−1 (O–H stretch), ca. 3020 cm−1 (C–H stretch in phenyl rings), ca. 2950 cm−1 (C–H stretch in aliphatic, i.e. CH2, groups), 1610 cm−1 (phenyl ring breathing mode), 1210 cm−1 (C–OH stretch), and 751 and 812 cm−1 (characteristic of ortho- and para-substituted phenolic groups).21,22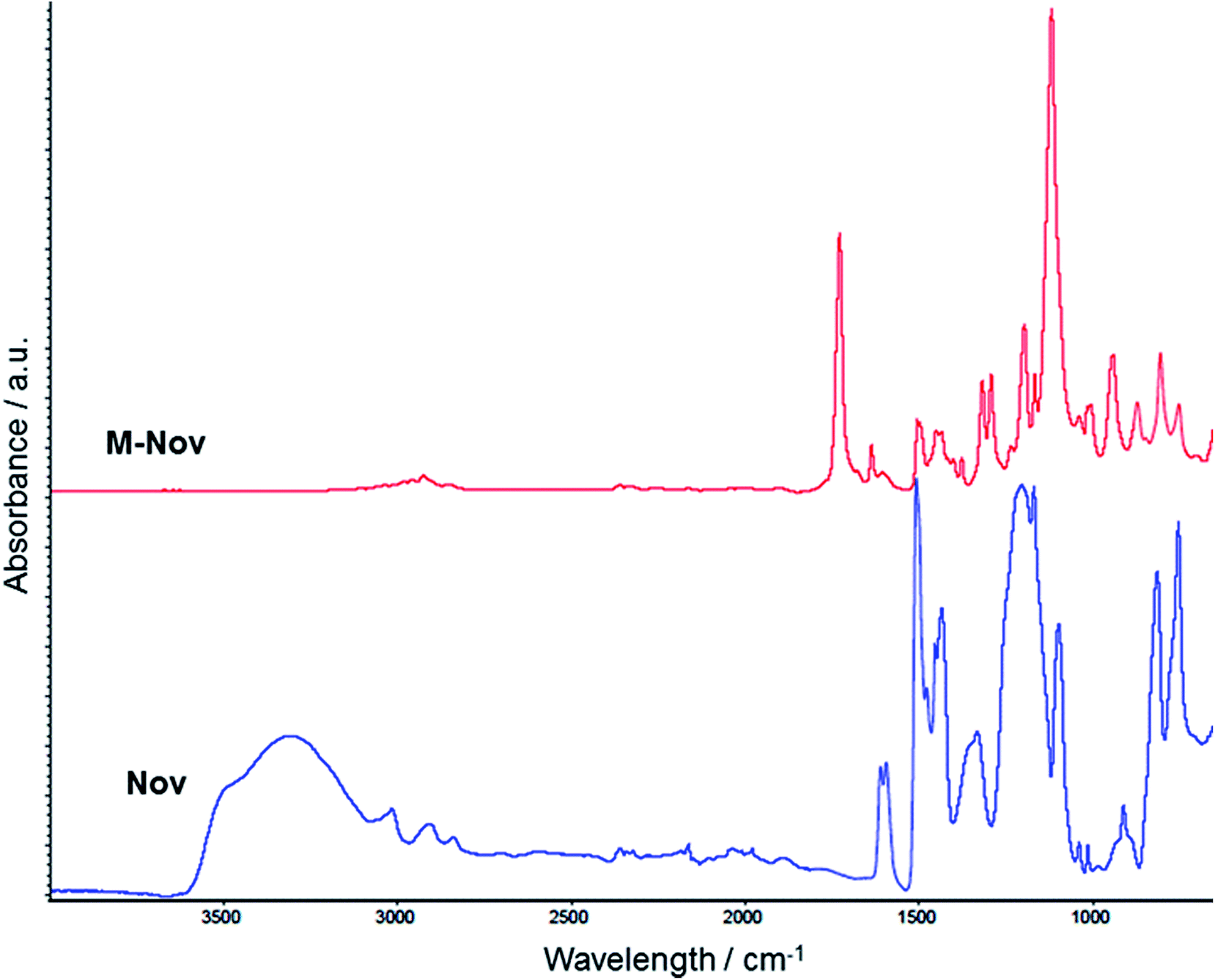 | ||
| Fig. 2 ATR spectra of Nov and M-Nov. | ||
The ATR spectrum of M-Nov provides clear evidence for the presence of methacrylate groups with characteristic peaks at 1730 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) and 1635 cm−1 (CC stretch).23,24 In addition, the disappearance of the hydroxyl peak at 3310 cm−1 in the spectrum of M-Nov indicates the complete reaction of phenolic OH with methacryloyl chloride.
O stretch) and 1635 cm−1 (CC stretch).23,24 In addition, the disappearance of the hydroxyl peak at 3310 cm−1 in the spectrum of M-Nov indicates the complete reaction of phenolic OH with methacryloyl chloride.
Proton NMR analysis of Nov and M-Nov
Proton NMR spectra of Nov and M-Nov are shown in Fig. 3. The spectrum of Nov contains peaks arising from phenolic OH protons (7.8–8.5 ppm), from protons in aromatic rings (6.5–7.1 ppm), and from protons in methylene links (3.4–3.8 ppm). The sharp signal at ca. 1.9 ppm arises from acetone solvent, whilst the broader peak at ca. 2.8 ppm may be from water. In the spectrum of M-Nov (Fig. 3), the peak from phenolic OH is absent, indicating that methacrylation of phenolic nuclei is complete. In addition, the spectrum of the methacrylated product contains two new broad signals of similar area between 5.4 and 6.2 ppm, which are assigned to the olefinic methylene protons of the methacrylate groups, and a broad signal centred at 1.8 ppm, which arises from the α-methyl protons of the methacrylate substituents. The breadth of these signals, like others in the spectrum, and like those in the spectrum of the parent Nov, are a consequence of the significant viscosity of the NMR solution. However, overlaid on these new broad signals in spectrum of M-Nov are two much sharper signals at 5.7 and 6.1 ppm. These also arise from olefinic protons, but are from a trace of unreacted methacryloyl chloride. There are also some sharp signals in the spectrum of M-Nov that can be assigned to solvents used in the work-up following synthesis. All the above assignments have been checked using an on-line NMR simulation package.25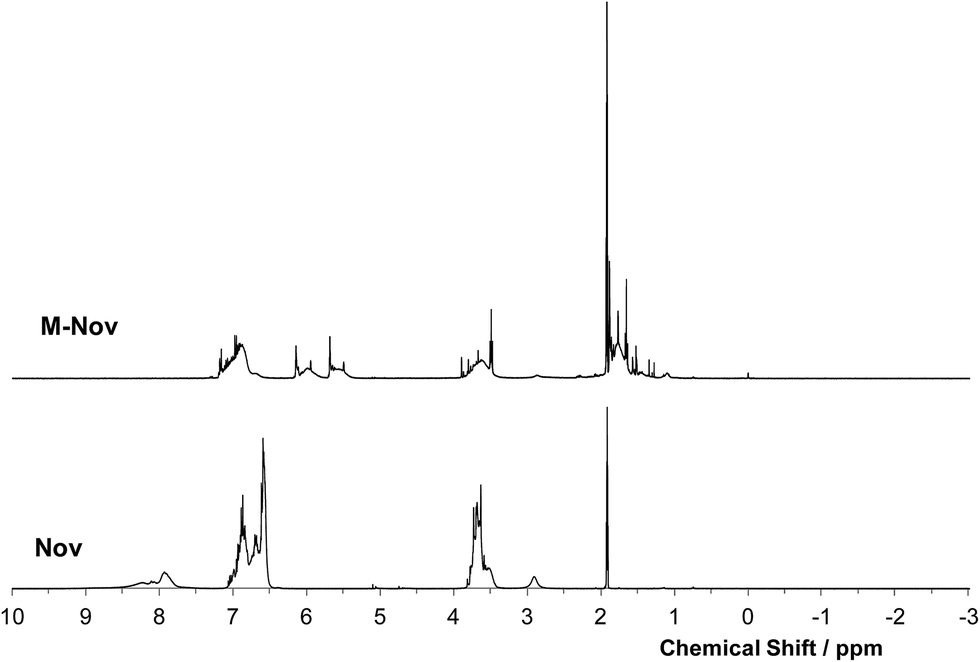 | ||
| Fig. 3 Proton NMR spectra of Nov and M-Nov. | ||
ATR FT-IR analysis of cured resins and resin blends
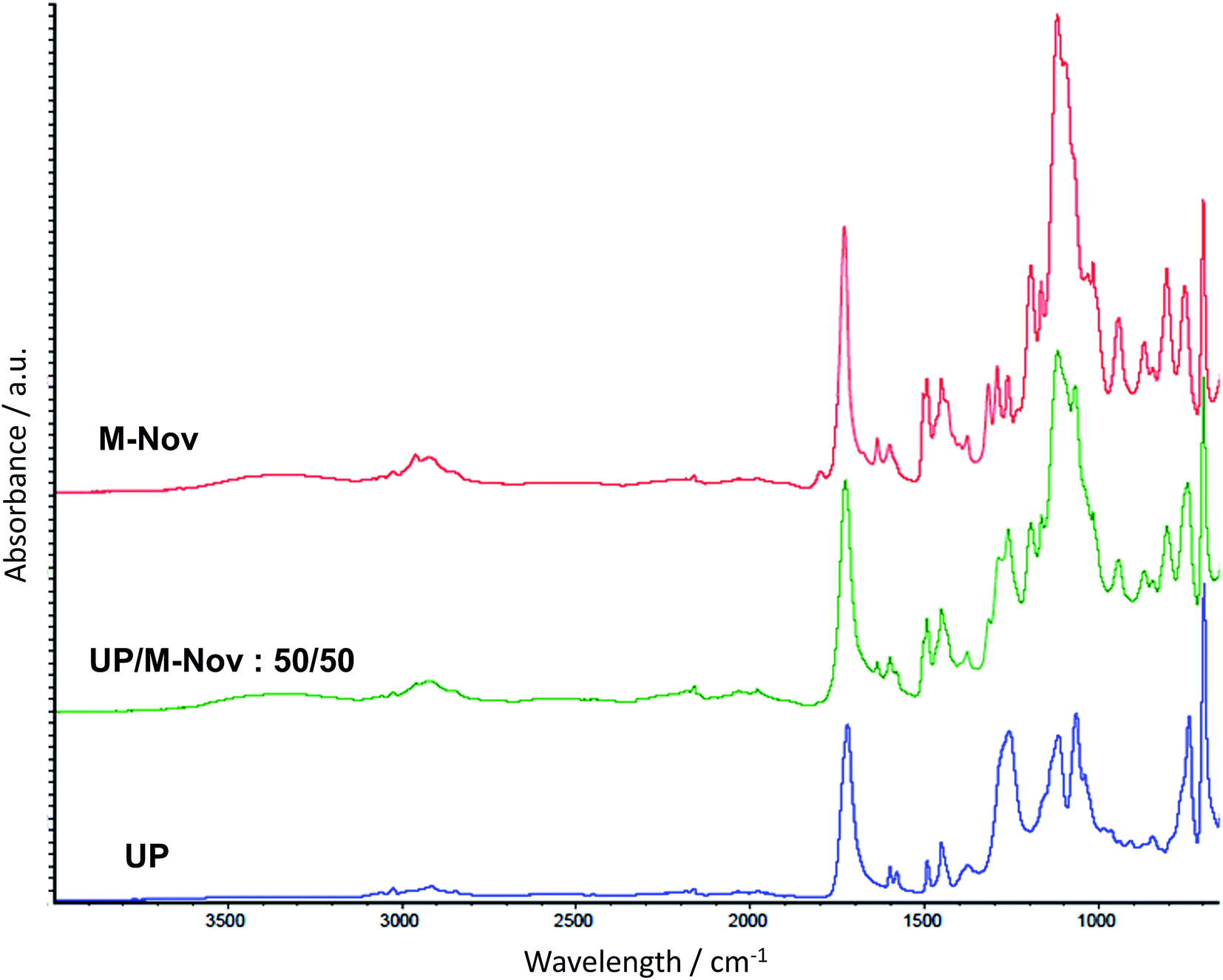
Fig. 4 shows the ATR spectra of cured cast samples of UP, M-Nov and a UP/M-Nov:50/50 blend. The spectrum of UP/Nov:50/50 exhibits, as expected, all the peaks of both UP and M-Nov, with the relative intensities of the bands roughly mirroring the relative proportions of the blend components, as expected. Although curing is extensive, there is evidence of the presence still in these resins after curing of small CC peaks at ca. 1635 cm−1. It is possible that these arise because some CC bonds in the UP and M-Nov become inaccessible to reaction as cure proceeds and the viscosity of the medium rapidly rises. It is not possible to determine from the spectra alone whether or not any chemical reaction takes place between the components of a blend during curing.
 | ||
| Fig. 4 ATR spectra of cured M-Nov, UP and UP/M-Nov:50/50 cast resins. | ||
Solid-state C13 NMR spectra of cured UP, M-Nov and UP/M-Nov blends
Solid-state C13 NMR spectra of cured UP, M-Nov and 50/50 and 70/30 cured blends of UP and M-Nov are shown, stacked, in Fig. 5. The most notable difference between the spectra is in the size of the peak centred at 68 ppm. This peak is absent in the spectrum of cured M-Nov and is at a maximum in the spectrum of UP, with the blends displaying peaks with intensities intermediate between those of the pure components. Spectral simulation25 shows that this peak arises only from the methine and methylene carbons of propylene glycol units in UP, thus it is to be expected that its intensity varies in proportion to the amount of UP in the blend. There are also significant differences in other regions of the spectra. For example, the signal from the phenolic carbon to which the methacrylate group is attached (ring C1) at 148 ppm diminishes in size as the amount of M-Nov in the resin is reduced, as expected. There are also more subtle changes in the spectra between 162 and 168 ppm (CO of maleate and phthalate groups in UP and of methacrylate groups in M-Nov), at 122 ppm (ring C2 at junction with CH2 link in M-Nov), between 32 and 56 ppm (methylene and methine carbons in polymerized maleate, methacrylate and styryl groups), and between 14 and 20 ppm where the methyl carbon signal of propylene glycol units in UP (ca. 16 ppm) is progressively replaced by the α-methyl carbon signal of methacrylate units (ca. 18 ppm) as the amount of UP in the blend is decreased and the amount of M-Nov is increased. Note that the spectra in Fig. 5 contain artifacts in the form of spinning side bands between 220 and 260 ppm with their mirror-image counterparts at 0–12 ppm and at 30 ppm.
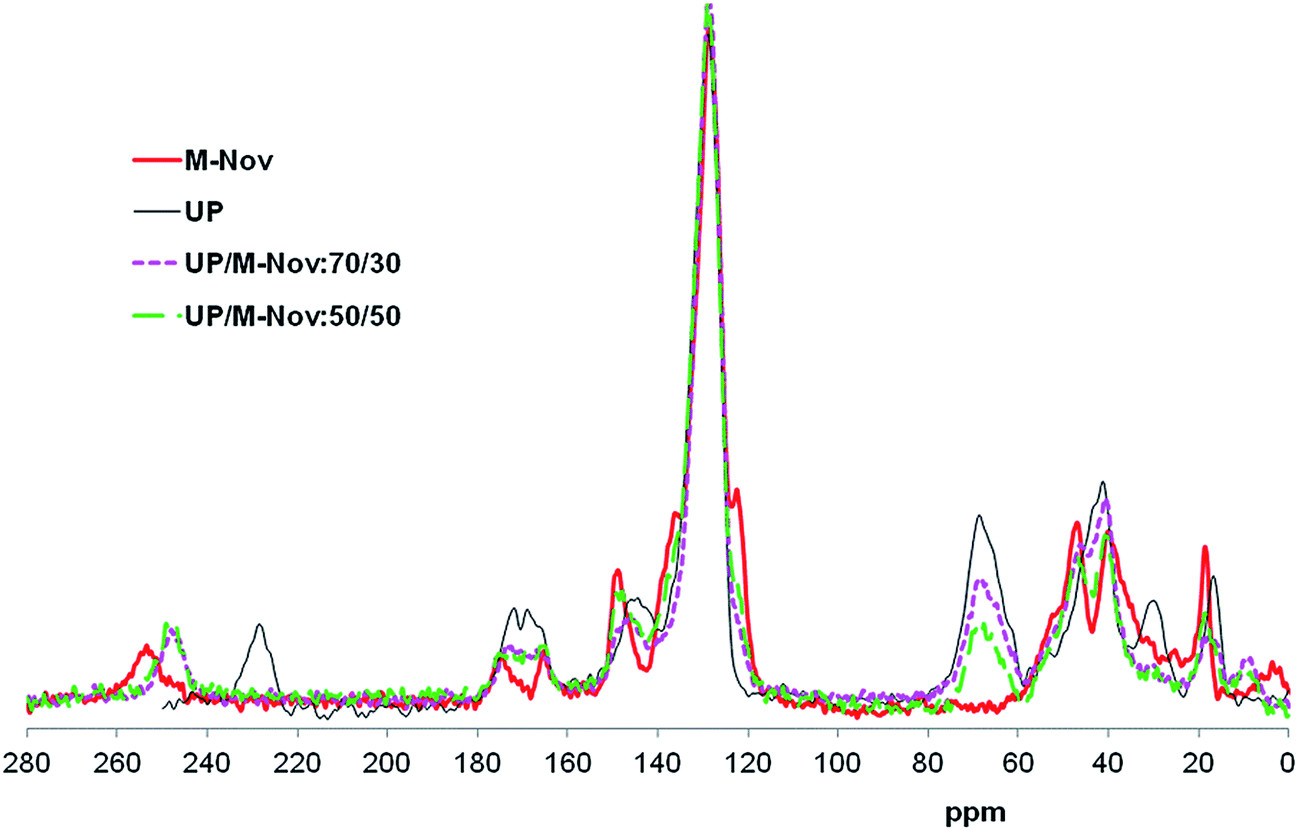 | ||
| Fig. 5 Solid-state C13 NMR spectra of cured UP, M-Nov and UP/M-Nov blends. | ||
Curing behaviour studied by DSC
M-Nov differs in its curing behaviour from Nov in that Nov is a thermoplastic polymer, and can only be cured by a high temperature condensation process after addition of a source of formaldehyde, such as hexamethylenetetramine.26,27 M-Nov, on the other hand, has been designed to cure, after addition of a reactive monomer such as styrene, by a relatively low-temperature free-radical process, similar to that used to cure UP/styrene mixtures.The DSC trace of uncured M-Nov, containing 32% by weight of styrene as the crosslinking monomer, plus Catalyst M, Accelerator G and dimethyl aniline as the initiator system, exhibits a curing exotherm similar to that of UP/styrene (Fig. 6(a)), as do those of the UP/M-Nov/styrene blends (Fig. 6(b)). This suggests that M-Nov/styrene cures in a manner analogous to UP/styrene, and that UP/M-Nov blends co-cure in a similar, single process. Reruns of the (now cured) samples on the DSC (also shown in Fig. 6(a) and (b)) give essentially flat traces, showing that curing is complete after the first programmed temperature run. The exotherm onset and peak temperatures, Tonset and Tpeak, and the areas under the exotherms (i.e. enthalpies of curing) for UP/styrene, M-Nov/styrene and the blends with styrene are listed in Table 1; the figures in parentheses are enthalpies of curing for blends calculated using eqn (1), assuming that these will be the mass averages of the values for the pure blend components. The enthalpy of curing for M-Nov/styrene is 222 J g−1 i.e. lower than that for UP/styrene (282 J g−1),10 similarly Tpeak for the M-Nov/styrene is 13 °C lower than that for UP/styrene; this latter feature may be a consequence of the expected greater reactivity in free-radical polymerization of the pendent, methacrylate, double bonds of M-Nov compared with the in-chain, olefinic, double bonds of the maleate units of UP. The data in Table 1 indicate that it is appropriate to cure M-Nov and blends of M-Nov with UP under conditions similar to those established for UP alone, i.e. 24 h at RT plus 6 h at 80 °C.10
| Pblend = (PUP × mUP) + (PM-Nov × mM-Nov) | (1) |
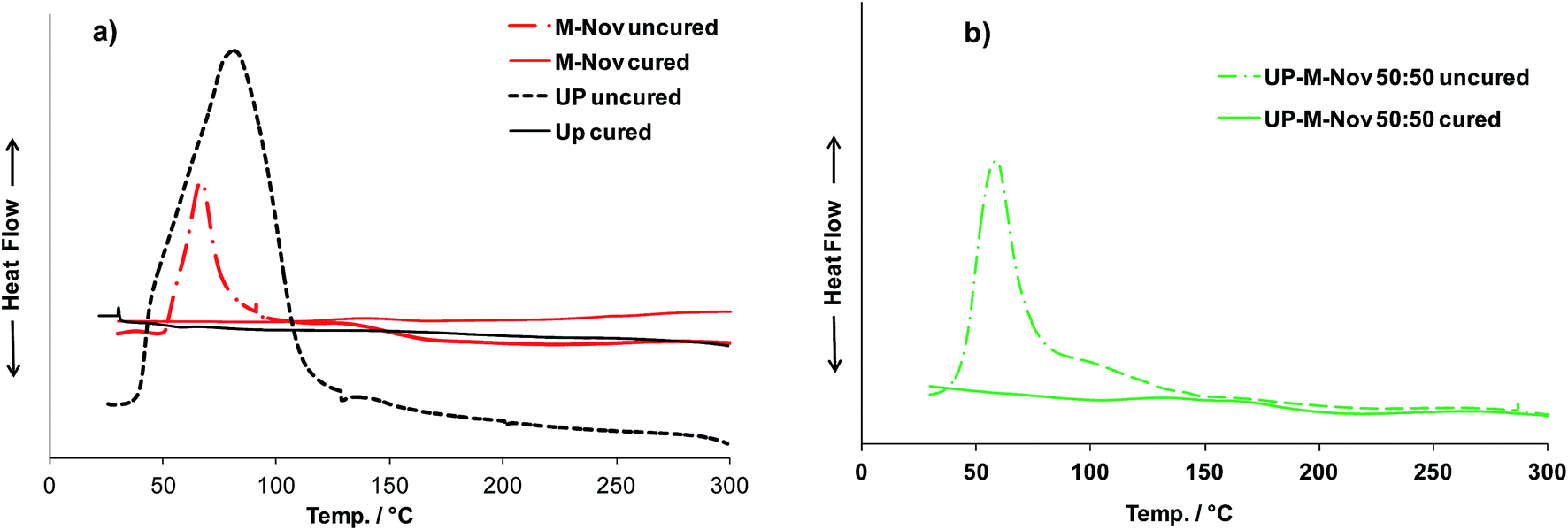 | ||
| Fig. 6 DSC curves of uncured and cured samples of (a) M-Nov and UP, (b) UP/M-Nov:50/50 resins. | ||
| Sample | DSC | DMTA | |||
|---|---|---|---|---|---|
| Tonset/°C | Tpeak/°C | Enthalpy of cure/J g−1 | Storage modulus at RT/MPa | Tg/°C | |
| a Note: the enthalpies in parentheses are mass-average values calculated from those of the components using eqn (1). | |||||
| M-Nov | 49 | 67 | 222 | 2130 | 182 |
| UP | 30 | 82 | 282 | 2657 | 92 |
| UP/M-Nov:80/20 | 35 | 60 | 271 (270) | 2860 | 116 |
| UP/M-Nov:70/30 | 36 | 55 | 262 (264) | 3250 | 123 |
| UP/M-Nov:60/40 | 36 | 60 | 266 (258) | 3011 | 125 |
| UP/M-Nov:50/50 | 44 | 59 | 258 (252) | 2591 | 126 |
Mechanical properties and resin compatibility studied by DMTA
DMTA was performed on all cast resin samples. From tanδ versus temperature curves, the glass transition temperature, Tg, for each sample was determined; the tanδ curves are shown in Fig. 7(a) and Tg values are reported in Table 1. The pure resins, UP and M-Nov, each display a tanδ curve with a single maximum corresponding to a single Tg, as expected. Similar to the other cured phenolic resins discussed in our earlier papers,9,10 the cured M-Nov resin has much higher Tg than that of UP.
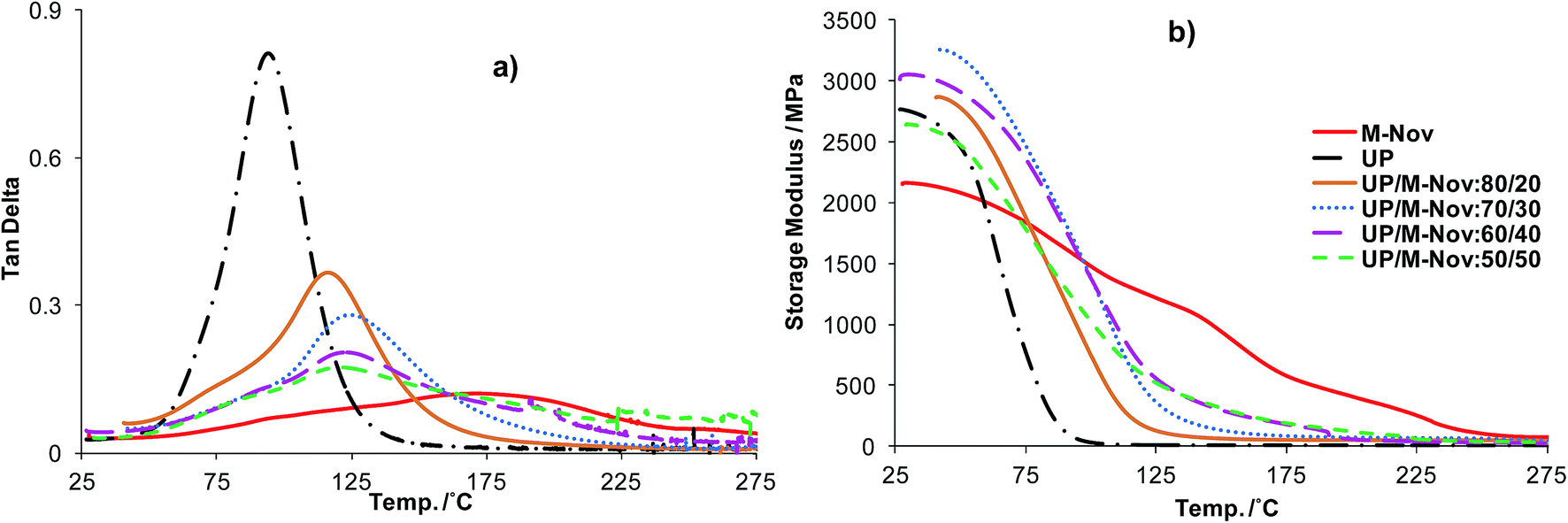 | ||
| Fig. 7 (a) tanδ and (b) storage modulus vs. temperature for cured M-Nov, UP and UP/M-Nov blends. | ||
The blends also show single tanδ vs. T maxima indicating single Tg values and hence good miscibility. This implies that the resin blends behave as single homogeneous materials (effectively terpolymers) and that M-Nov can be co-cured to form a single continuous network with UP and styrene. As expected, the Tg values of the blends are between those of the individual resins (92 °C for UP and 182 °C for M-Nov), and increase as the amount of M-Nov in the blend is increased. However, the Tgs of the 70/30, 60/40 and 50/50 blends of UP/M-Nov are unexpectedly similar and not in accordance with the Fox equation, from which we would expect the Tg of a blend to be clearly dependent upon the weight fractions of the components.28 This may be due to differences in crosslink density and/or to slight compositional heterogeneity, as discussed below.
The overall compositions of these network blends will be the same as the compositions of the mixtures from which they have been made, given that polymerization and crosslinking is taken to maximum conversion. However, the compositions will undoubtedly drift during the polymerization and crosslinking process owing to expected differences in the reactivity ratios of the component species. Reactivity ratios of M-Nov and UP with each other and with other monomers are not known, but if we take diethylmaleate (DEM) to be a model for the polymerizable groups in UP and methyl methacrylate (MMA) to be a model for M-Nov, then the relevant reactivity ratio pairs have been reported to be: MMA/DEM = 354/0;29 MMA/styrene = 0.50/0.54;30 styrene/DEM = 6.592/0.001.31 From these pairs we can see that we would expect M-Nov to be reacted into the network much more readily than UP, giving a partial product richer in M-Nov at the outset of the polymerization and crosslinking process. This could lead to microscale heterogeneity within the fully crosslinked network but, if so, this heterogeneity is clearly not sufficient to cause significant phase separation and hence the observation of more than one Tg.
Also obtainable from the DMTA data are the storage moduli of the cured resins and resin blends; these are shown as a function of T in Fig. 7(b), with values at RT (25 °C) listed in Table 1. The variation of the storage modulus with temperature is mainly used to assess the likely retention of mechanical strength at high temperature. A material displaying a relatively slow decrease in modulus with increasing temperature will perform better at higher temperatures than one in which the modulus declines sharply.
In these respects, it can be seen from Table 1 and Fig. 7(b) that cured M-Nov has a modulus lower than that of cured UP at RT (indicating that it is a softer material than cured UP at this temperature) but retains this modulus more effectively at higher temperatures than does UP. Particularly noteworthy are the performances of the cured 80/20, 70/30 and 60/40 UP/M-Nov blends, all of which show superior storage moduli over the whole temperature range compared with that of cured UP, and indeed higher moduli than that of cured M-Nov. The reason for this latter feature is not clear but may, like the trend in Tgs, be a reflection of different extents of curing of the various resins.
Morphology of cured cast resins of UP, M-Nov and UP/M-Nov studied by SEM
The DMTA data indicate good compatibility between UP and M-Nov resins. To further assess compatibility, the surface morphologies of fractured sections of cured cast resin samples were examined by SEM. Fig. 8 shows the SEM images of the fractured surfaces of the cured resins. The fracture surface of cured M-Nov is slightly rougher than that of UP. The blended resins also display slightly rougher surfaces than fractured UP but with no voids and no obvious signs of phase separation, such as those we have observed with some UP/resole blends.10 Most probably, this also indicates blend homogeneity, and the existence within the blend of a fully co-crosslinked polymer network. | ||
| Fig. 8 SEM photomicrographs of fractured surfaces of cured M-Nov, UP and 70/30 and 50/50 UP/M-Nov blends. | ||
Limiting oxygen indices (LOI)
The effect of M-Nov on the ease of ignition of UP in the UP/M-Nov blends was determined by measurement of LOI. The measured LOI values are listed in Table 2 together with mass-average values (in italics) calculated using eqn (1). It can be seen that the LOIs of the cured blends lie between those of the pure components, as expected, and that they do not differ greatly from the calculated values. We take this to be also an indication of blend homogeneity. Of more importance in this context of course is that cured M-Nov, and cured blends of M-Nov with UP, have better LOI than cured UP.| Sample | LOI/vol% O2 | ΔLOI (increase over that of UP) |
|---|---|---|
| a Note: the LOI values in italics are calculated mass-average values using eqn (1). | ||
| M-Nov | 21.3 ± 0.1 | 3.4 |
| UP | 17.9 ± 0.1 | 0 |
| UP/M-Nov:80/20 | 19.0 ± 0.1 (18.6) | 1.1 |
| UP/M-Nov:70/30 | 19.1 ± 0.2 (18.9) | 1.2 |
| UP/M-Nov:60/40 | 19.5 ± 0.1 (19.3) | 1.6 |
| UP/M-Nov:50/50 | 19.7 ± 0.1 (19.6) | 1.8 |
Cone calorimetric assessment of fire retardance of cured resins and resin blends
The cone parameters, TTI, FO, PHRR, THR, TSR and CY for UP, M-Nov and their blends, are listed in Table 3 with FPI and FIGRA values calculated from these. The percentage reduction in different parameters, ΔPHRR (PHRR of sample minus PHRR of UP), ΔTHR and ΔTSR are also given in Table 3 to illustrate the effect of M-Nov content in the blends. From Table 3, it can be seen that the time to ignition of M-Nov (41 s) is similar to that of UP. However, M-Nov resin burns for a longer time (FO time 202 s, compared to 178 s for UP). The performances of the blends are intermediate between those of UP and M-Nov in these respects. The PHRR in M-Nov is 29% lower than that of UP (see Table 3). Moreover, the total heat released during burning of M-Nov (61 MJ m−2) is 29% less than that in UP (83 MJ m−2). The char yield in M-Nov is 19.3% whilst in UP it is only 1.9%. In M-Nov, the char acts as thermal barrier similar to other phenolic resins.4,11 However, the char yield of cured M-Nov is lower than those of unmodified novolac resins27 because of the absence of combustible methacryloyl groups and styrene crosslinks in the latter.| Sample | TTI/s | FO/s | PHRR/kW m−2 | THR/MJ m−2 | TSR/m2 m−2 | CY/% | FPI/kW−1 m2 s | FIGRA/kW m−2 s−1 |
|---|---|---|---|---|---|---|---|---|
| a Notes: (1) the probable errors for the cone parameters are: TTI ± 2; PHRR ± 33; THR ± 2.1; TSR ± 133 and CY ± 1.3. (2) FPI = TTI/PHRR; FIGRA = PHRR/TPHRR. (3) The negative numbers in parentheses are the percentage reductions in values with respect to those for UP. | ||||||||
| M-Nov | 41 | 202 | 801 (−29) | 61 (−27) | 3512 (−27) | 19.7 | 0.051 | 8.09 |
| UP | 38 | 178 | 1130 | 83 | 4813 | 1.9 | 0.034 | 10.09 |
| UP/M-Nov:80/20 | 39 | 178 | 988 (−13) | 70 (−16) | 4103 (−15) | 5.7 | 0.039 | 10.74 |
| UP/M-Nov:70/30 | 39 | 181 | 932 (−18) | 68 (−18) | 3909 (−19) | 7.6 | 0.042 | 9.23 |
| UP/M-Nov:60/40 | 38 | 183 | 892 (−21) | 66 (−21) | 3815 (−21) | 9.7 | 0.043 | 9.39 |
| UP/M-Nov:50/50 | 39 | 187 | 840 (−26) | 64 (−23) | 3623 (−25) | 12.4 | 0.046 | 8.66 |
The PHRR, THR, TSR and CY values for the UP/M-Nov 80/20, 70/30, 60/40 and 50/50 blends all lie between those of the pure components, as expected. However, the relationships between PHRR, THR, TSR and blend composition are not linear ones, with the incorporation of even small relatively amounts of M-Nov in UP having a disproportionately advantageous effect on these parameters. A possible reason for this feature is presented and discussed in a later section.
It is interesting to compare the cone calorimetric parameters for the UP/M-Nov blends with those recorded previously for blends of UP with Methylon (an allyl-functional resole that was found to co-cure to a limited extent with UP/styrene) of similar composition.4 For UP/Methylon:50/50, for example, PHRR was 800 kW m−2, THR was 61 MJ m−2, TSR was 3170 m2 m−2 and char yield was 14% w/w; for UP/M-Nov:50/50, the corresponding data are 840 kW m−2, 64 MJ m−2, 3623 m2 m−2 and 12.4% w/w, i.e. very similar to the results for the UP/allyl-resole blend. There is a similar close correspondence too for the data for the two 50/50 blends. Thus, from the point of view of their flame retardance, UP/M-Nov blends perform as well as UP/Methylon blends. This is particularly encouraging given that, prior to curing, extra styrene was added to the UP/M-Nov blends to give an overall concentration of styrene in the blended resin of about 35% w/w, whereas for the UP/Methylon blends, no extra styrene is added over that which is already present in the UP. So, for example, the UP/Methylon:50/50 blend contains only about 18% w/w styrene (a particularly flammable component).
The UP/M-Nov blends are also ranked with two other fire parameters, FPI and FIGRA, listed in Table 3. The fire performance index, FPI, is derived from TTI/PHRR. The fire growth rate, FIGRA is maximum quotient of HRR(t)/TPHRR, which often equals to PHRR/TPHRR in a cone calorimeter. From the point of view of flame retardancy, materials with higher FPI are preferred over materials with lower FPI because of the greater fire risks associated with the latter. From Table 3, it can be seen that the FPI values of UP/M-Nov blends increase with increasing M-Nov content, indicating that UP/M-Nov blends pose a lower fire risk than UP.
FIGRA is associated with the burning propensity of a material. From Table 3 it can be seen also that the FIGRA values of UP/M-Nov blends are lower than that of UP, indicating slower fire growth in the blends once ignition has occurred.
Thermal and thermo-oxidative stabilities of cured resins and resin blends
Thermal stabilities of cured UP, M-Nov and UP:M-Nov blends have been investigated by thermogravimetric (TGA) and differential thermal analysis (DTA), under both nitrogen and air. Fig. 9(a1) and (a2), presents the mass loss vs. T plots for UP, M-Nov and their blends in nitrogen and air, respectively, whilst Fig. 9(b1) and (b2) are the corresponding derivative (DTG, i.e. rate of mass loss vs. T) plots, and Fig. 9(c1) and (c2) the corresponding DTA plots. Salient parameters derived from these plots are listed in Tables 4 and 5.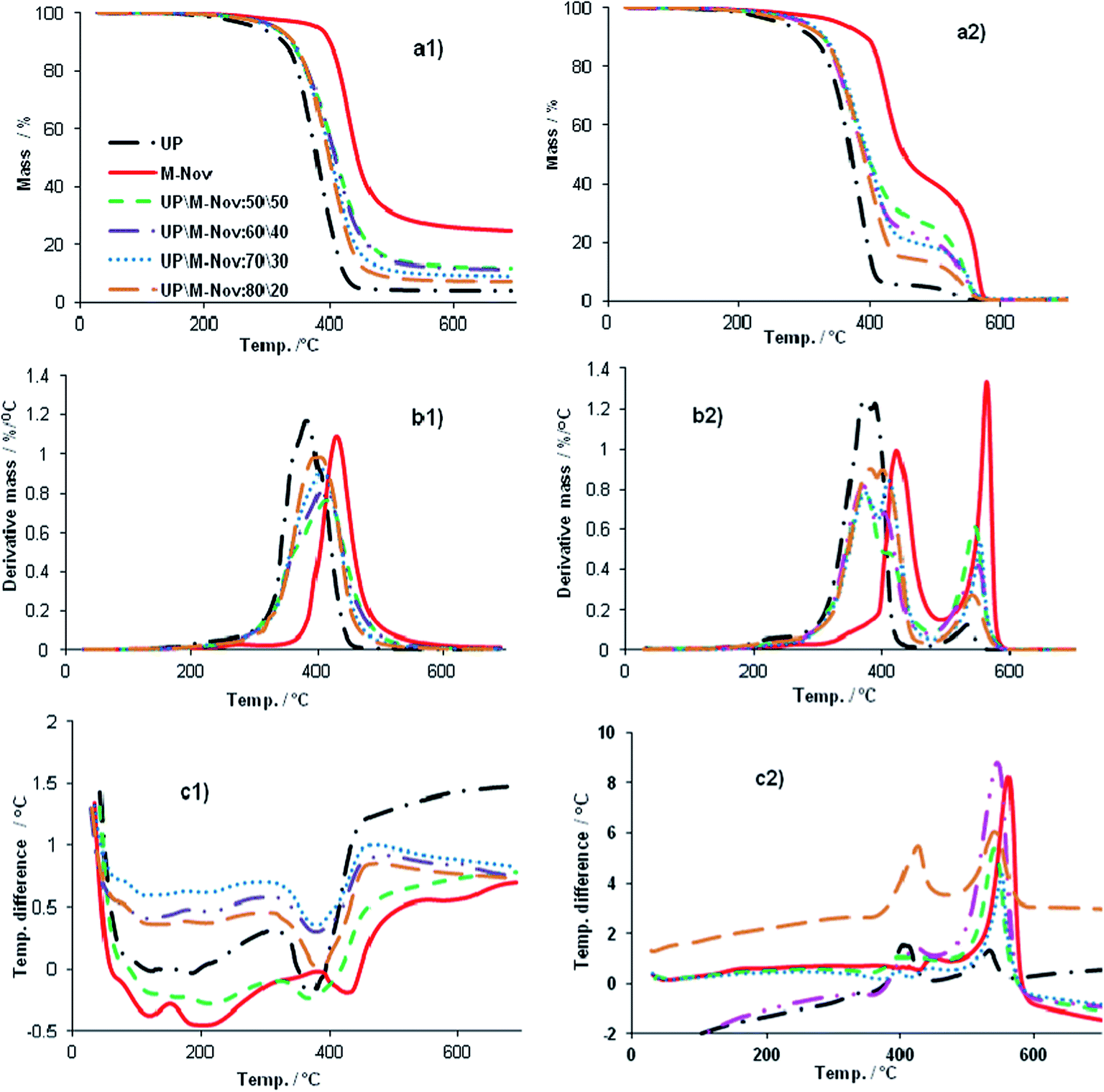 | ||
| Fig. 9 TGA traces in nitrogen (a1) and air (a2), DTG traces in nitrogen (b1) and air (b2), and DTA traces in nitrogen (c1) and air (c2) for cured M-Nov, UP and UP/M-Nov blends. | ||
| Sample | Temp. range/°C | Mass loss/% | DTG max/°C | DTA peak max/°C | Residue at 525 °C/% |
|---|---|---|---|---|---|
| M-Nov | RT–246 | 0.9 | 431 | 152 (exo) | 28.7 |
| 246–567 | 72.3 | 432 (endo) | |||
| UP | RT–183 | 0.9 | 383 | 2.5 | |
| 183–462 | 94.8 | 369 (endo) | |||
| UP/M-Nov:80/20 | RT–206 | 0.7 | 403 | 174 (exo) | 7.9 |
| 206–503 | 90.8 | 386 (endo) | |||
| UP/M-Nov:70/30 | RT–213 | 0.8 | 406 | 174 (exo) | 10.2 |
| 213–516 | 88.8 | 380 (endo) | |||
| UP/M-Nov:60/40 | RT–223 | 0.7 | 411 | 174 (exo) | 12.9 |
| 223–516 | 86.7 | 380 (endo) | |||
| UP/M-Nov:50/50 | RT–231 | 1.1 | 414 | 173 (exo) | 13.7 |
| 231–537 | 85.6 | 371 (endo) |
| Sample | Temp. range/°C | Mass loss/% | DTG max/°C | DTA peak max/°C | Residue at 525 °C/% |
|---|---|---|---|---|---|
| a Note: s = small shoulder peak. | |||||
| M-Nov | RT–295 | 2.7 | 35.3 | ||
| 295–489 | 55.7 | 422 | 427 (endo) | ||
| 489–601 | 41.2 | 563 | 561 (exo) | ||
| UP | RT–183 | 0.9 | 2.8 | ||
| 183–435 | 93.1 | 373 | 352 (endo):404 (exo) | ||
| 435–566 | 5.6 | 532 | 533 (exo) | ||
| UP/M-Nov:80/20 | RT–206 | 1.4 | 355 (endo) | 9.5 | |
| 206–459 | 84.5 | 381 | 425 (exo) | ||
| 459–599 | 14.1 | 540 | 538 (exo) | ||
| UP/M-Nov:70/30 | RT–251 | 2.1 | 377 (endo) | 14.8 | |
| 251–472 | 78.3 | 371, 411 (s) | 397 (exo), 441 (s, exo) | ||
| 472–593 | 19.2 | 522 | 553 (exo) | ||
| UP/M-Nov:60/40 | RT–251 | 2.3 | 368 (endo) | 15.2 | |
| 251–475 | 75.3 | 370, 404 (S) | 391 (s, exo), 423 (exo) | ||
| 475–596 | 22.1 | 548 | 545 (exo) | ||
| UP/M-Nov:50/50 | RT–251 | 2.4 | 348 (endo) | 19.1 | |
| 251–477 | 70.7 | 372, 413 (S) | 392 (exo) | ||
| 477–594 | 26.7 | 543 | 541 (exo) | ||
It can be seen from Fig. 9(a1) and (b1), and Table 4 that cured M-Nov exhibits a single stage mass loss under nitrogen between 246 °C and 567 °C, with peak rate (DTG max) at 431 °C, leaving a residual mass of 28.7% at 525 °C. Associated with this mass loss is an endothermic DTA peak at 432 °C arising from the volatilization of the degradation products. The small exothermic DTA peak at 152 °C probably arises from some slight further curing of M-Nov during the DTA run. This overall behaviour of M-Nov is similar to that of UP, but cured M-Nov is significantly more thermally stable than cured UP, which degrades almost completely in a single stage between 183 and 462 °C, leaving a residue of only 2.5%. The cured UP/M-Nov blends also show single stage mass loss over temperature ranges intermediate between those observed for cured M-Nov and cured UP e.g. 90.8% mass loss between 206 and 503 °C for UP/M-Nov:80/20 and 85.6% mass loss between 231 and 537 °C for UP/M-Nov:50/50. We take this pattern of behaviour to indicate further the homogeneous nature of the blends, i.e. they behave as single polymeric materials with uniform chemical composition throughout. This behaviour is in marked contrast to that which we previously observed for cured blends of UP with some commercial phenolic resoles; these thermally decomposed in two or more stages indicating significant domains of pure UP and pure phenolic in phase-separated structures.4 The small mass losses that can be seen for the various samples between RT and the onset of the major degradation process (e.g. 0.9% for M-Nov and 1.1% for UP/M-Nov:50/50) almost certainly arise from the volatilization of small amounts of solvent, residual monomer and other low molar mass fragments in the materials.
When thermal degradation of the samples is carried out in air, two major regions of mass loss are seen for all the samples (see Fig. 9(a2), (b2) and (c2) and Table 5). The first region in each case (e.g. between 295 and 489 °C for M-Nov and between 251 and 477 °C for UP/M-Nov:50/50) corresponds to thermal and thermo-oxidative degradation of the original polymer, whilst the second (e.g. between 489 and 601 °C for M-Nov, and between 477 and 594 °C for UP/M-Nov:50/50) corresponds to the thermo-oxidative degradation of the otherwise thermally stable residue formed during the first degradation process (what can be termed a char oxidation stage). Both these regions of mass loss in all but pure M-Nov are accompanied by significant exothermic DTA peaks (Fig. 9(c2)), characteristic of exothermic oxidation processes (e.g. at 392 and 541 °C for UP/M-Nov:50/50, 397 and 553 °C for UP/M-Nov:70/30, and 404 and 533 °C for UP). These exotherms overwhelm the endothermic peaks that would be expected from volatilization of degradation products. Only in M-Nov is a single exotherm seen, probably indicating the greater oxidative stability of M-Nov and the products of its thermal decomposition compared with those of UP, leading to the exothermic peak corresponding to oxidation matching in size the endothermic peak corresponding to volatilization of degradation products.
Comparison of the residual masses for the various samples under nitrogen and air at 525 °C is also informative. Whereas in UP the residual mass at 525 °C in air is only marginally greater than in nitrogen (2.8% vs. 2.5%), for M-Nov the difference is much more marked (35.3% in air and 28.7% in nitrogen). The residual mass differences in air and nitrogen for the blends are intermediate between those of UP and M-Nov. This too is an indication of the greater thermo-oxidative stability of M-Nov compared with UP in that the residue remaining after the major stage of thermal decomposition initially gains mass through oxidation (replacement of H by O) before finally oxidatively degrading above 525 °C; in all samples, no mass remains in air above 600 °C.
In order to see whether or not blends of M-Nov and UP are more or less thermally and thermo-oxidatively stable than might be expected on the basis of their compositions and the behaviours of pure M-Nov and UP, mass-averaged mass loss curves have been calculated for the blends using eqn (1). The differences between experimental mass losses and calculated mass losses for all blends are plotted against T in Fig. 10(a) and (b).
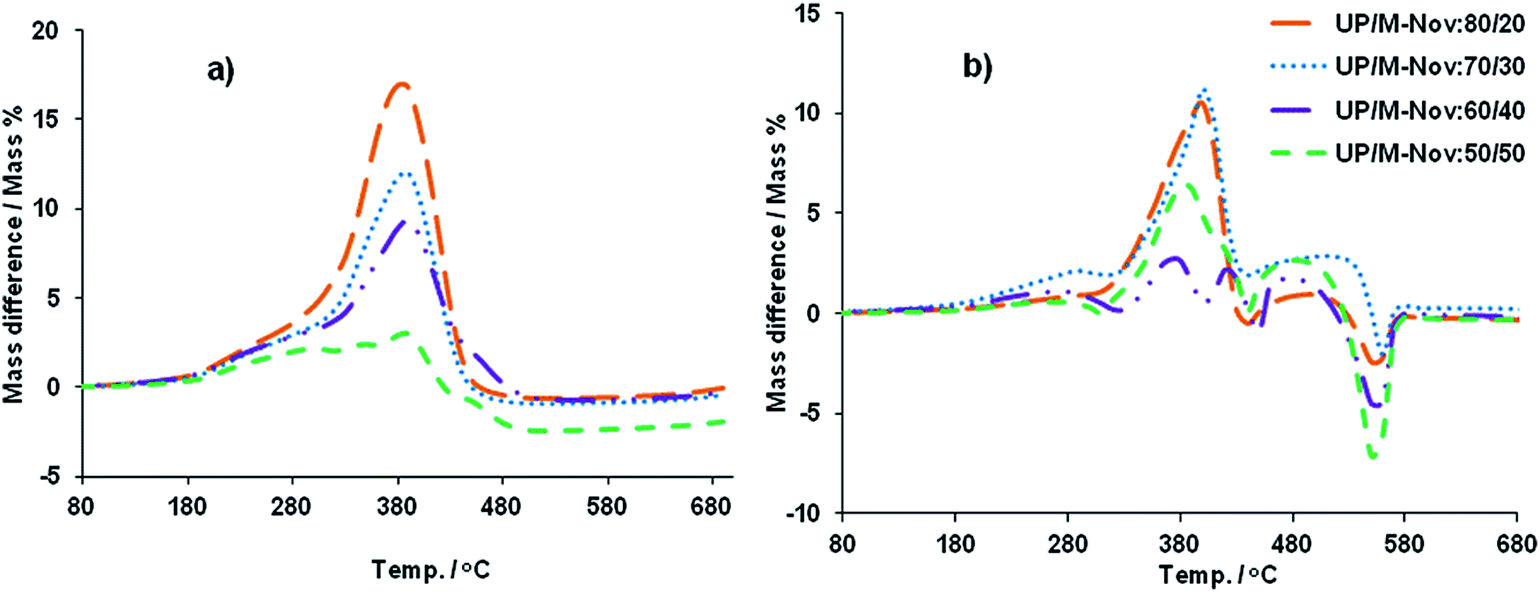 | ||
| Fig. 10 Mass differences between experimental and calculated mass loss curves vs. T for all UP/M-Nov blends in (a) air and (b) nitrogen. | ||
From Fig. 10(a) and (b), it can be seen that in both nitrogen and air, the blends are more stable (lose less mass) than expected on the basis of eqn (1) between about 180 and 530 °C. This indicates that the resins are truly co-cured and have formed a co-continuous network structure in which the sequences of phenolic units have a thermally protective effect on the interwoven polyester sequences. Moreover, this increase in thermal and thermo-oxidative stability over that which might reasonably have been expected, i.e. lower than expected amounts of volatiles released, especially if flammable, is consistent with the increased flame retardancy of the blends (i.e. reduced PHRR and THR) beyond that which would be expected on the basis of their compositions, as seen in the data presented in Table 3, and discussed above. The release of volatiles from degrading M-Nov, UP and UP/M-Nov blends has been studied in more detail by FTIR evolved gas analysis, and the results of this are discussed in the next section.
Evolved gas analysis using TGA-FTIR
We have discussed the application of TGA-FTIR to the analysis of gases evolved when cured phenolic resoles, UP and blends of UP with phenolic resoles are heated in an earlier paper,4 so will not go into great detail again here. Suffice it to say that the major products that can be identified in the gas stream emanating when a phenolic resin is pyrolysed under nitrogen, together with the IR bands used for quantification are: phenol (3647 cm−1); methane (3016 cm−1); CO2 (2360 cm−1); all compounds containing aliphatic C–H (2925 cm−1) and all compounds containing aromatic groups (3025 cm−1 and 1600 cm−1). The only difference in the results for the resole resins reported earlier4 and for the M-Nov seen here is that styrene is also evolved in the latter, which is due to the use of styrene as a cross-linker in M-Nov. For the unsaturated polyester resin, we can quantify: phthalic anhydride (1866 cm−1); and styrene (700 cm−1); plus CO2, total aliphatic species and total aromatic species, as with the phenolic resin.The IR spectra were recorded at temperatures between room temperature and 900 °C on gases being evolved from cured M-Nov, UP and a UP/M-Nov:70/30 blend. In Fig. 11, the absorbances of the IR bands arising from the gaseous degradation products of interest are plotted as a function of temperature for pure cured M-Nov, UP and UP/M-Nov blends. From these plots, total amounts of gases evolved (in arbitrary units) over the whole degradation temperature range can be determined by measuring the areas under the absorbance vs. T curves; these date are given in Table 6.
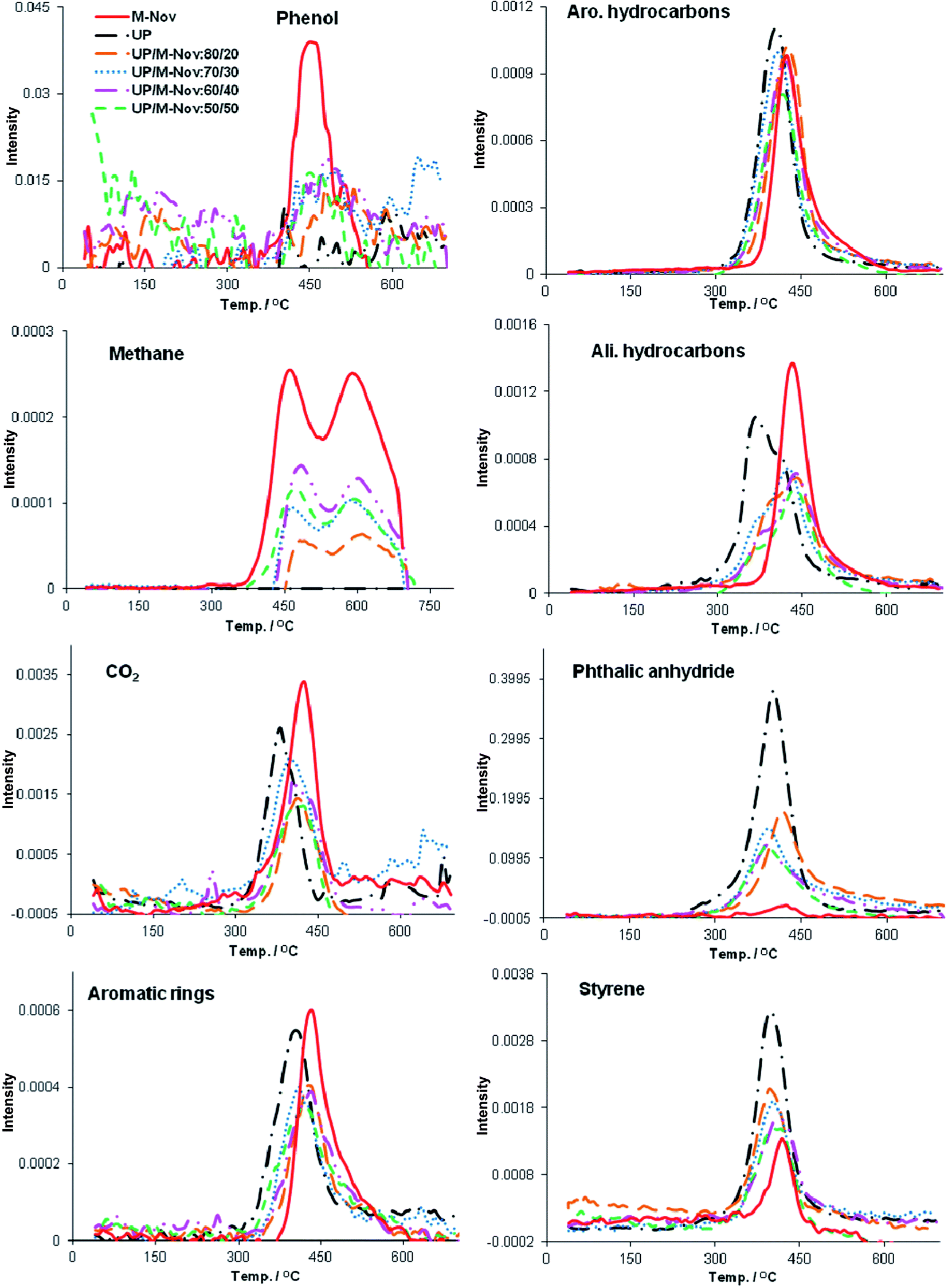 | ||
| Fig. 11 Absorbances of the IR bands characteristic of the various gaseous volatile pyrolysis products for M-Nov, UP and UP/M-Nov blends as a function of temperature. | ||
| Sample | Gas evolved (FTIR peak area × 10−2) | |||||||
|---|---|---|---|---|---|---|---|---|
| Gas | CO2 | Phenol | Phth. anhyd. | Styrene | Methane | Arom. rings | Arom. CH | Ali. CH |
| IR cm−1 | 2360 | 3647 | 1866 | 700 | 3016 | 1600 | 3025 | 2925 |
| a Note: the numbers in italics are mass-average amounts of gases expected from the blends, again calculated using eqn (1). | ||||||||
| M-Nov | 2.28 | 0.37 | 0.00 | 0.71 | 0.58 | 0.48 | 0.75 | 1.03 |
| UP | 1.91 | 0.00 | 2.68 | 2.53 | 0.00 | 0.39 | 1.11 | 1.49 |
| UP/M-Nov:80/20 | 1.58 | 0.07 | 1.52 | 1.88 | 0.12 | 0.31 | 0.86 | 0.89 |
| (1.98) | (0.07) | (2.14) | (2.17) | (0.12) | (0.41) | (1.04) | (1.40) | |
| UP/M-Nov:70/30 | 1.66 | 0.10 | 1.39 | 1.85 | 0.21 | 0.36 | 0.86 | 0.88 |
| (2.02) | (0.11) | (1.88) | (1.98) | (0.17) | (0.42) | (1.00) | (1.35) | |
| UP/M-Nov:60/40 | 1.71 | 0.12 | 1.26 | 1.59 | 0.27 | 0.42 | 0.86 | 0.84 |
| (2.06) | (0.15) | (1.61) | (1.80) | (0.23) | (0.43) | (0.97) | (1.30) | |
| UP/M-Nov:50/50 | 1.67 | 0.12 | 1.06 | 1.22 | 0.29 | 0.45 | 0.72 | 0.63 |
| (2.10) | (0.19) | (1.34) | (1.62) | (0.29) | (0.44) | (0.93) | (1.26) | |
It can be seen from Table 6 that the amounts of the various gaseous products evolved from the UP/M-Nov blends are intermediate between those of UP and M-Nov, as expected. However, calculation of amounts of gases that might be expected from the blends based on blend composition and using eqn (1) to calculate expected mass losses (i.e. eqn (1)) gives some values significantly different from those measured. In particular, yields of CO2 (evolved by both components), phthalic anhydride (only from UP), styrene (from both components) and total aliphatic species (from both components) are all lower than might be expected over the range of blend compositions studied. This is further evidence that the blends pyrolyse slightly less readily than might be expected and that this is possibly owing to some protection of the polyester sequences by the co-cured phenolic sequences, especially given that phthalic anhydride is only evolved by UP, whereas the yields of phenol and methane, which are evolved only from the UP, are not much different from the calculated values.
Conclusions
A methacrylated phenolic novolac resin (M-Nov) has been prepared that can be cured (crosslinked) free radically with styrene in a manner similar to that used with unsaturated polyesters (UP) to give a material with a higher Tg, better thermal and thermo-oxidative stabilities, and better flame retardancy than cured UP, albeit with an approximately 20% lower modulus at RT. Moreover, M-Nov can be mixed and co-cured free radically with UP over a range of compositions to give homogeneous materials (single Tg) with properties intermediate between those of cured M-Nov and cured UP. The flame retardancies of co-cured blends of M-Nov and UP are better than expected, arising, we believe, from some thermal protection of the relatively thermally unstable polyester sequences in the blend afforded by the methylene linked phenolic sequences. In separate experiments we have shown that glass-fibre reinforced composites having good mechanical, thermal and flame retardant properties can be made using M-Nov and M-Nov/UP blends, and we shall be reporting on this work separately.Acknowledgements
We thank the UK Engineering and Physical Sciences Research Council (EPSRC) for funding this work (Grant no. EP/H020675/1), Scott-Bader and Sumitomo-Bakelite Europe N.V. for provision of materials and facilities, and Dean Bugg, Jan Schreurs and Tom De Smedt of the aforementioned companies for technical advice. We thank also the University of Manchester, Department of Chemistry, for providing proton NMR spectra, and the EPSRC solid-state NMR service at the University of Durham for providing the solid-state C13 NMR spectra.References
- L. H. Baekeland, US-PS 942699, 1909.
- L. Pilato, React. Funct. Polym., 2013, 73, 270–277 CrossRef CAS PubMed.
- B. K. Kandola, A. R. Horrocks, P. Myler and D. Blair, in Fire and Polymers, ed. G. L. Nelson and C. A. Wilkie, ACS Symp Ser, 2001, vol. 799, pp. 344–360 Search PubMed.
- B. K. Kandola, L. Krishnan and J. R. Ebdon, Polym. Degrad. Stab., 2015, 113, 154–167 CrossRef CAS PubMed.
- J.-C. Munoz, H. Ku, F. Cardona and D. Rogers, J. Mater. Process. Technol., 2008, 202, 486–492 CrossRef CAS PubMed.
- A. Knop and L. A. Pilato, in Phenolic resins, Springer, Berlin, 1985 Search PubMed.
- R. Rego, P. J. Adriaensens, R. A. Carleer and J. M. Gelan, Polymer, 2004, 45, 33–38 CrossRef CAS PubMed.
- B. Ottenbourgs, P. Adriaensens, R. Carleer, D. Vanderzande and J. Gelan, Polymer, 1998, 39(22), 5293–5300 CrossRef CAS.
- B. K. Kandola, D. Deli and J. R. Ebdon, Compatibilised polymer blends, UK Patent Application, GB1222468.9, 2014.
- D. Deli, B. K. Kandola, J. R. Ebdon and L. Krishnan, J. Mater. Sci., 2013, 48, 6929–6942 CrossRef CAS.
- B. K. Kandola and L. Krishnan, Proceedings of the 11th International Symposium on Fire Safety Science (IAFSS), Christchurch, New Zealand, 10–14th Feb, 2014 Search PubMed.
- B. K. Kandola, L. Krishnan, D. Deli and J. R. Ebdon, Polym. Degrad. Stab., 2014, 106, 129–137 CrossRef CAS PubMed.
- C. S. Tyberg, K. Bergeron, M. Sankarapandian, P. Shih, A. C. Loos, D. A. Dillard, J. E. McGrath, J. S. Riffle and U. Sorathia, Polymer, 2000, 41, 5053–5062 CrossRef CAS.
- H.-T. Chiu and J.-O. Cheng, Polym.-Plast. Technol. Eng., 2007, 46, 801–810 CrossRef CAS.
- A. B. Cherian and E. T. Thachil, J. Appl. Polym. Sci., 2006, 100, 457–465 CrossRef CAS PubMed.
- F. Fekete, P. J. Keenan and W. J. Plant, Polyhydroxy polyacrylate esters of epoxidized phenol–formaldehyde novolac resins and laminates therefrom, US Pat. 3301743, 1967.
- P. Löcker, in Radiation curing: coatings and printing inks; technical basics, application and trouble shooting, Vincentz Network GmbH and Co., Section 3.3.2.1, 2008 Search PubMed.
- J. P. Godschalx, E. P. Woo, P. A. Schrader and P. D. Aldrich, Vinylbenzyl ethers of polyhydric halogenated phenolic compounds, European Patent 0258695 A1, 1991.
- C. Gouri, C. P. Reghunadhan Nair and R. J. Ramaswamy, J. Appl. Polym. Sci., 1999, 73, 695–705 CrossRef CAS.
- B. Biswas and B. K. Kandola, Polym. Adv. Technol., 2011, 22, 1192–1204 CrossRef CAS PubMed.
- Z. Y. Wang, Y. Liu and Q. Wang, Polym. Degrad. Stab., 2010, 95, 945–954 CrossRef CAS PubMed.
- L. Cui, S. Wang, Y. Zhang and Y. Zhang, J. Appl. Polym. Sci., 2007, 104, 3337–3346 CrossRef CAS PubMed.
- H. Liang, A. Asif and W. Shi, Polym. Degrad. Stab., 2005, 87, 495–501 CrossRef CAS PubMed.
- B. Karagoz and N. Bicak, Eur. Polym. J., 2008, 44, 106–112 CrossRef CAS PubMed.
- Nmrdb.org, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Federale de Lausanne, France, December 2014, http://www.nmrdb.org.
- A. Artmann, O. Bianchi, M. R. Soares and C. R. Regina, Mater. Sci. Eng., 2010, C30, 1245–1251 CrossRef PubMed.
- M. J. Sumner, M. Sankarapandian, J. E. McGrath, S. Riffle and U. Sorathia, Polymer, 2002, 43, 5069–5076 CrossRef CAS.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123–128 CAS.
- W. I. Bengough, D. Goldrich and R. A. Young, Eur. Polym. J., 1967, 3, 117–123 CrossRef CAS.
- V. E. Meyer, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 2819–2830 CrossRef CAS PubMed.
- D. Braun and G. Cei, Makromol. Chem., 1986, 187, 1713–1726 CrossRef CAS PubMed.
Footnote |
| † Present address: Romer Labs UK Ltd, The Heath Business & Technical Park, Runcorn, WA7 4QX, UK. |
| This journal is © The Royal Society of Chemistry 2015 |