
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective oxidation of uronic acids into aldaric acids over gold catalyst†
Sari
Rautiainen
a,
Petra
Lehtinen
a,
Jingjing
Chen
a,
Marko
Vehkamäki
a,
Klaus
Niemelä
b,
Markku
Leskelä
a and
Timo
Repo
*a
aLaboratory of Inorganic Chemistry, Department of Chemistry, University of Helsinki, A. I. Virtasen aukio 1, P.O. Box 55, 00014 University of Helsinki, Finland. E-mail: timo.repo@helsinki.fi; Tel: +358-2941-50194
bVTT-Technical Research Centre of Finland, P.O. Box 1000, 02044 VTT, Finland
First published on 9th February 2015
Abstract
Herein, uronic acids available from hemicelluloses and pectin were used as raw material for the synthesis of aldaric acids. Au/Al2O3 catalyst oxidized glucuronic and galacturonic acids quantitatively to the corresponding glucaric and galactaric acids at pH 8–10 and 40–60 °C with oxygen as oxidant. The pH has a significant effect on the initial reaction rate as well as desorption of acid from the catalyst surface. At pH 10, a TOF value close to 8000 h−1 was measured for glucuronic acid oxidation. The apparent activation energy Ea for glucuronic acid oxidation is dependent on the pH which can be attributed to the higher energy barrier for desorption of acids at lower pH.
Introduction
Carbohydrates are abundant raw materials for the sustainable production of chemicals.1,2 Aldaric acids, diacids of sugars, are important building block chemicals3 and have numerous applications in polymer, detergent and pharmaceutical industries, but their production methods are limited.4–6 Traditionally, aldaric acids are produced by oxidation of saccharides with nitric acid in ∼40% yields.7 The method has been further improved in terms of both yield and acid recovery,8,9 and recently, commercial pilot-scale production of glucarate-products by oxidation of D-glucose was started by Rivertop Renewables.10 Alternative oxidation pathways have also been studied. Nitroxide-mediated oxidation of glucose using bleach and NaBr gives glucaric acid in >90% yield.11,12 From green chemistry point of view, however, it would be more sustainable to use catalytic methods employing air, molecular oxygen or hydrogen peroxide.13 Due to their multifunctionality, the oxidation of carbohydrates can result in multiple products; e.g. the aerobic, platinum-catalysed oxidation of monosaccharides gives only 50–66% of aldaric acids.14,15 Development of selective catalyst for carbohydrate oxidation is therefore required.The aldehyde group of monosaccharides is efficiently oxidized to aldonic acids using supported gold nanoparticles (Au NPs).16 These catalysts have shown superior selectivity and stability in comparison to other noble metal catalysts.17–19 Mildly alkaline conditions (pH range 8–10) are required to neutralize the produced acid and keep the catalyst surface active. Metal oxide supported Au NPs, e.g. Au/Al2O3, have shown very high selectivity and long-term stability in oxidation of glucose to gluconic acid.20 Other aldoses are also selectively oxidized; however, the effect of carbohydrate structure on reactivity is not quite clear.16
Oxidation of the primary hydroxyl group of monosaccharides to the aldaric acid requires more severe conditions and often results in lower selectivity.21 However, in case of uronic acids the primary hydroxyl is oxidized already by nature, which makes them highly attractive raw material for the production of bio-based chemicals. Glucuronic and galacturonic acids are components of hemicelluloses in both hardwood and softwood and can amount up to 6% of wood dry weight.22 To date, hemicelluloses are underutilized and thus very appealing polysaccharides for wood-based biorefineries. Galacturonic acid is also the main constituent of pectin, which is available in abundance from e.g. citrus peels.23 In addition, substantial amounts of mannuronic and guluronic acids are available from alginates.24
In general, there are few examples of the oxidation of uronic acids to aldaric acids reported in literature, including use of stoichiometric oxidants such as manganese(III) sulfate25 or arylhaloamines.26 Additionally, galactarate can be produced from galacturonic acid with high yields through a biotechnological process, though the incubation times were long.27
In our preliminary studies for oxidation of uronic acids using Au supported on Al2O3, TiO2 and MgO, the best results were obtained with Au/Al2O3 catalyst.28 This type of catalyst, prepared by direct ion-exchange, was previously reported effective also in the oxidation of arabinose and galactose.29,30 Our aim was to broaden the substrate scope as well as to gain insight on the effect of carbohydrate structure on the catalyst activity. While continuing our studies, van der Klis et al. reported selective oxidation of galacturonic acid using commercial gold catalysts, mainly Au/TiO2.31 In augmentation of the previous reports, we show herein the quantitative and highly efficient oxidation of D-glucuronic and D-galacturonic acids into the corresponding aldaric acids using Au/Al2O3 and oxygen as oxidant (Scheme 1).
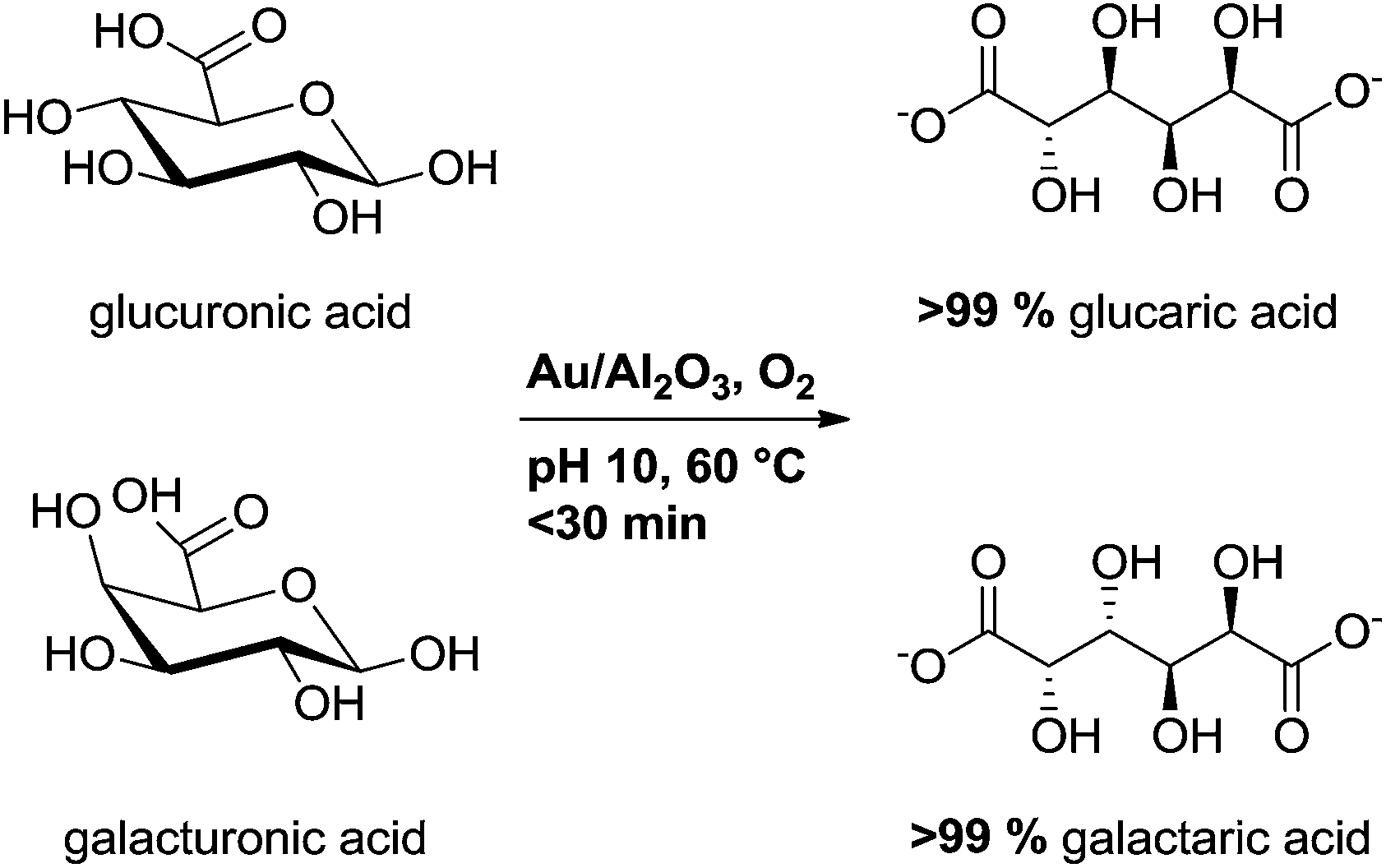 | ||
| Scheme 1 Oxidation of uronic acids into corresponding aldaric acids. | ||
Experimental
Catalyst preparation
The Au/Al2O3 catalyst was prepared using direct ion exchange method.32 A 5 × 10−4 M aqueous solution of HAuCl4 (99.9%, ABCR) was prepared corresponding to final Au loading of 2 wt%. The solution was heated to 70 °C and powdered Al2O3 (Alfa Aesar, γ-phase, 40 μm, S.A. 200 m2 g−1) was added. The slurry was mixed for 1 hour, washed with 100 ml 4 M ammonia for 1 hour, filtered, dried overnight at 70 °C and calcined in air at 300 °C for 4 h.Catalyst characterization
Gold particle size distribution and the metal dispersion were determined by transmission electron microscopy (TEM) by counting 100 particles from multiple separate catalyst particles. A FEI Tecnai F20 TEM operated at 200 kV was used for collecting the bright-field images. A catalase crystal standard was used for scale calibration. According to the result, the average Au particle size was 2.4 ± 0.6 nm (Fig. 1). Gold dispersion DAu was 43%, calculated from the particle size distribution as reported by Delidovich et al.33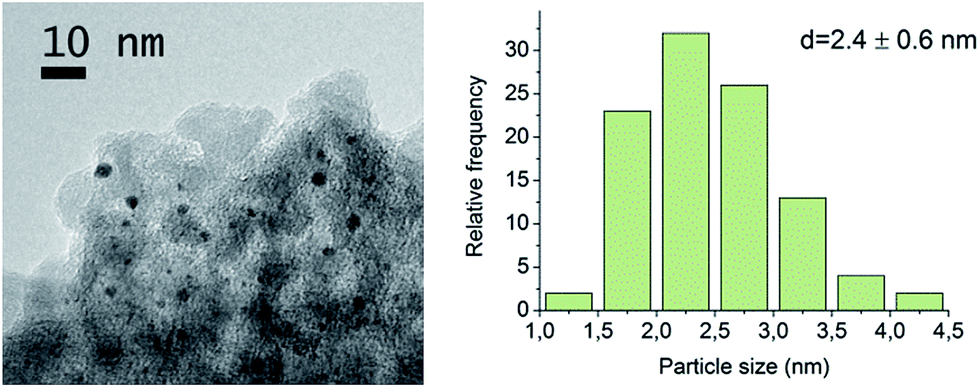 | ||
| Fig. 1 TEM image and gold particle size distribution of Au/Al2O3. | ||
Gold loading was determined by AAS (Atomic Absorption Spectrophotometer, PerkinElmer 3030) after dissolving the catalyst into aqua regia. The fresh Au/Al2O3 catalyst contained 1.8 ± 0.15 wt% Au.
Oxidation procedure
D-glucuronic acid (GlcA, ≥98%, Sigma) and D-galacturonic acid monohydrate (GalA, ≥98%, Sigma) were used as such. An automatic titrator (Metrohm Titrando 907) filled with 1 M aqueous NaOH was used to maintain constant pH during the reaction. In a typical experiment, uronic acid (e.g. 2.58 mmol), 25 mg Au/Al2O3 catalyst (0.09 mol% Au compared to the substrate) and 50 ml deionized water were measured in a three-necked round-bottom flask fitted with a condenser and pH electrode (Metrohm Unitrode). The mixture was stirred with a magnet at 1400 rpm and heated to the desired temperature. At the start of the reaction the carboxylic acid group of the uronic acid was neutralized and the solution titrated to the set pH. Dioxygen was bubbled at 100 ml min−1 through a gas dispersion tube (Sigma-Aldrich) to the reaction mixture. The reaction was stopped when the addition of NaOH ceased, indicating the completion of the reaction. Catalyst and substrate amounts were optimized so that oxygen solubility was not a limiting factor for the reaction. Reproducibility of the results was confirmed at pH 10 and 60 °C, 3% deviation was detected in the activity in two repeated experiments. In control experiments, no reactions occurred with only the alumina support or without the catalyst.To isolate glucaric acid by precipitation of its monopotassium salt, glucuronic acid was oxidized with 0.05 mol% Au/Al2O3 at pH 10 using 1 M KOH as the titrant. After the oxidation, the filtered solution was cooled to 20 °C and pH was adjusted to 3.4 using nitric acid.7 The white precipitate was filtered, washed with a small amount of cold water and dried at 60 °C, resulting in 72% yield.
Product analysis
The products were identified by 1H-NMR, 13C-NMR spectroscopic methods and GC/MS analyses (cf. ESI†). Selectivity was determined using 1H-NMR spectroscopy and HPLC. Due to the very high selectivity of oxidation (>99%) to aldaric acid, conversions can be determined from NaOH consumption, as the addition of base corresponds directly to the acid formation. NMR spectra were measured directly from the reaction solution using presaturation technique to suppress the solvent water signal. For GC/MS analysis, the sample was cation-exchanged to H+, dried and trimethylsilylated.34Evaluation of reaction rate
Conversion vs. time plots were derived from the NaOH consumption. Gas flow was started only when the correct pH and temperature were reached, resulting in a short induction period during which the solution was saturated with oxygen. Therefore, specific activities (mmol per gAu per min) were calculated from the linear part of the kinetic curve, between conversions 10–50%. Turn-over frequencies (TOF) per surface Au atom were calculated from the conversion after 15 min reaction according to| TOF = nsubstratec/nAuDAut |
Results and discussion
Uronic acids and carbohydrates are sensitive towards degradation and isomerization in highly alkaline media, and pH values over 10 should be avoided.35–37 Consequently, the oxidation reactions were carried out using an automatic titrator to maintain constant pH and to avoid the use of excess base. At the start of the reaction, the uronic acid was neutralized and pH set to the desired value (e.g. pH 10).The oxidation of uronic acids was carried out using oxygen bubbling at 100 ml min−1. The addition of base during the reaction corresponds to the formation of the second carboxylic acid in the product; the reaction progress can be followed from the NaOH consumption. 1H-NMR confirmed the conversion determined from NaOH consumption. With 0.09 mol% Au/Al2O3 at pH 10 and room temperature, glucuronic acid (GlcA) was selectively oxidized to glucaric acid with 95% conversion in 2 h (Table 1, entry 1). Increasing the temperature led to considerable increase in the reaction rate, and at 60 °C full conversion was achieved in 23 min reaching TOF value close to 8000 h−1 (Fig. 2 and Table 1, entry 2). Analysis of the reaction solution by 1H-NMR, 13C-NMR and GC/MS revealed the quantitative formation of glucarate (see ESI†). The controlled continuous addition of base shown herein is favourable in terms of selectivity; no isomerization, degradation or side reactions were observed. In comparison, the oxidation of glucuronic acid with Au/TiO2 and 1 equiv. NaOH, added at the beginning of the reaction, gives lower glucarate yield (85%) even in 5 h.31
| Entry | Substrate | pH | T (°C) | Specific activity (mmol per gAu per min) | TOF (h−1) | Selectivity (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 2.58 mmol substrate, 0.09 mol% Au, 50 ml water, 100 ml min−1 O2. Conversion >99% unless otherwise stated. b Conversion 95%. c Catalyst recycled by washing with water and drying. TOF not calculated due to changed dispersion. | ||||||
| 1b | GlcA | 10 | 25 | 103 | 2270 | >99 |
| 2 | GlcA | 10 | 60 | 297 | 7920 | >99 |
| 3 | GlcA | 9 | 60 | 238 | 5970 | >99 |
| 4 | GlcA | 8 | 60 | 130 | 3250 | >99 |
| 5 | GalA | 10 | 60 | 286 | 6840 | >99 |
| 6 | Glc | 10 | 60 | 391 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
>99 |
| 7 | Gal | 10 | 60 | 276 | 6900 | >99 |
| 8c | GlcA | 10 | 60 | 191 | n/a | >99 |
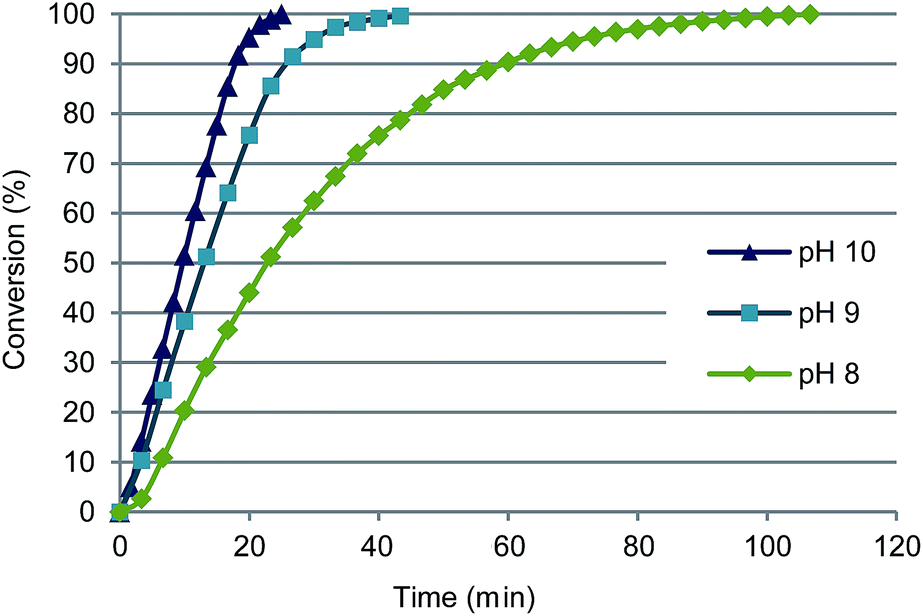 | ||
| Fig. 2 Effect of pH on oxidation of glucuronic acid with 0.09 mol% Au/Al2O3 at 60 °C. Conversions were calculated from the consumption of base (NaOH) using an automatic titrator. | ||
The pH value has a strong effect on the activity of gold catalysts, and this was observed also in GlcA oxidation with Au/Al2O3. At pH 8–9 GlcA was also quantitatively converted to glucaric acid, though the initial reaction rate decreased with decreasing pH (Table 1, entries 3 and 4 and Fig. 2). At pH 10, Au/Al2O3 maintained the initial activity until 85% conversion, while at pH 8 the rate started decreasing already after 40% conversion. High conversion (94%) was reached also at neutral pH 7, although the reaction time was prolonged (4 h). The decrease of the initial oxidation rate can be explained by adsorption of free acid on the catalyst surface, consequently blocking the active sites.38 At low pH, small part of the carboxylic acid groups of the substrate and product are protonated, as the produced acid is not completely neutralized. In alkaline medium however, the acids are deprotonated and readily desorb the catalyst surface.
Further studies on the effect of reaction conditions on the oxidation were carried out at 40–70 °C and pH 8–10. Under these conditions, both temperature and pH had a favourable effect on the catalyst activity (Fig. 3). The highest activity 370 mmol per gAu per min was observed at 70 °C and pH 10, however, the selectivity dropped to 98%. Generally, temperatures over 60 °C should be avoided in carbohydrate oxidation to prevent side reactions.36 Interestingly, the temperature increase had stronger effect on the oxidation at pH 8 compared to pH 9–10.
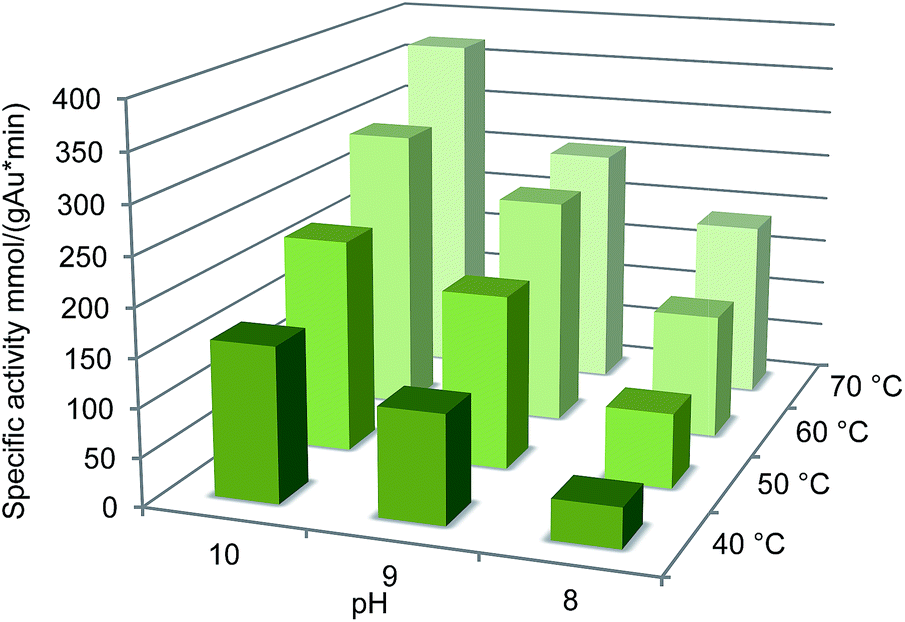 | ||
| Fig. 3 Effect of pH on oxidation of glucuronic acid with 0.09 mol% Au/Al2O3 at 60 °C. Conversions were calculated from the consumption of base (NaOH) using an automatic titrator. | ||
Based on the experimental data, the apparent activation energies Ea were calculated for the oxidation at each pH using Arrhenius plots (Fig. S9†). In heterogeneous catalysis, the apparent activation energy observed is the activation energy of the reaction modified by heat of adsorption ΔHad of the reaction species.39 Accordingly, the activation energy of a reaction does not depend on the concentration of the reactants. At pH 10, the apparent activation energy for glucuronic acid oxidation was 24.6 kJ mol−1, and at pH 9 a slightly higher value, 26.5 kJ mol−1 was obtained. However, at pH 8 Ea increased to 45.3 kJ mol−1. These values are comparable to those reported for oxidation of glucose (27–47 kJ mol−1)40,41 and arabinose (23.8 kJ mol−1).42 In these previous studies however, the effect of pH on Ea has not been addressed. As discussed above, pH influences the deprotonation and desorption of acids. At pH 8, the acids are not completely deprotonated and adsorb more strongly to the catalyst surface compared to pH 9–10. Consequently, the increased apparent activation energy may be attributed to the higher energy barrier of acid desorption.
The pH value influences greatly the initial reaction rate. At the start of the reaction the substrate is neutralized and product concentration is low; catalyst deactivation should not yet affect the reaction rate. This suggests that hydroxide ions participate in the oxidation itself. It has been proposed that the hydroxide ion reacts with the aldehyde group of the carbohydrate and form an open chain hydrate which is then dehydrogenated on the catalyst surface.38,43 Thus, the oxygen atoms in the produced carboxylic acid originate from hydroxides in water, not dioxygen, as was shown by isotopic labeling studies on oxidation of 5-hydroxymethylfurfural.44 Dioxygen completes the catalytic redox cycle by scavenging electrons on the catalyst surface and is consequently reduced to peroxides and further to hydroxide.45
The GlcA isomer, galacturonic acid (GalA) was oxidized quantitatively into galactaric acid using Au/Al2O3 with activity 286 mmol per gAu per min and TOF value 6840 h−1 (Table 1, entry 5). The two substrates differ only by the stereochemistry of C4 and no marked difference in catalytic activity was observed (Scheme 1). To further study the influence of the carbohydrate structure on the catalysis, the oxidation of GlcA and GalA was compared to their corresponding hexoses, D-glucose (Glc) and D-galactose (Gal). Of the four substrates studied herein, the highest activity was observed in the oxidation of Glc to gluconic acid. The catalytic activity is 30% higher than in GlcA oxidation (Table 1, entries 2 and 6). This comparison indicates that the carboxylate at C6 has a rate-decreasing effect. In addition, Gal having an axial hydroxyl at C4 oxidizes to galactonic acid with 30% lower catalytic activity than Glc and, moreover, with similar activity as the abovementioned GalA and GlcA (entries 2, 5 and 7). Accordingly, the axial hydroxyl at C4 and the carboxylate at C6 have similar rate decreasing effect on the oxidation. Unexpectedly, these effects are not cumulative; GalA, although having both axial hydroxyl at C4 and the carboxylate at C6, oxidizes with Au/Al2O3 as easily as Gal and GlcA.
In previous studies with Au/TiO2, Mirescu et al. observed similar rate-decreasing effect of the axial hydroxyl at C4; Glc and Gal were oxidized with specific activities 56 and 34 mmol per gAu per min, respectively.16 In contrast, De Wit et al. observed different behaviour for Pt/C; the oxidation of Gal proceeded considerably faster than Glc and also, the oxidation rate of GlcA and GalA decreased 80% compared to their corresponding hexoses.46 The oxidation with Pt/C was proposed to proceed through dehydrogenation of the cyclic carbohydrate to an intermediate lactone which is then hydrolysed to the acid. Clearly, the different behaviour of the two metals supports the proposed different oxidation mechanisms.
The reusability of Au/Al2O3 was studied by washing and drying the catalyst after GlcA oxidation at pH 10. In a successive reaction, the very high selectivity was maintained, though the activity dropped (entry 8). Possible causes for catalyst deactivation include metal leaching, gold particles sintering, poisoning and adsorption of reactants or products on the catalyst surface. Depending on the strength of the adsorption, the adsorbed molecules could be removed by washing or calcination. Several washing treatments were applied to remove possible weakly adsorbed molecules from the catalyst surface (Fig. S10†). However, neither base nor acid wash improved the activity of the spent catalyst compared to water wash. Also, catalyst calcination and thermogravimetric analysis (TGA, see ESI†) did not show considerable mass loss, indicating that adsorption is not the cause of the deactivation. Gold leaching was studied by determining the gold content of the catalyst as well as the reaction solution. Both the fresh and spent Au/Al2O3 contained similar amount of Au, within the experimental error of AAS. Correspondingly, no leached gold was detected in the reaction solution. Finally, the spent Au/Al2O3 catalyst was studied by TEM. In Fig. 4, aggregation and coalescence of gold particles is clearly visible indicating that the major cause of the deactivation is the reduced Au surface area. The size of the aggregates ranges from 6.0 to 9.5 nm, while some smaller particles are also present. However, accurate determination of the average particle size and dispersion is problematic due to the irregular shape of the particles.
 | ||
| Fig. 4 TEM image of spent Au/Al2O3 after oxidation of glucuronic acid. | ||
Conclusions
We have shown that selective oxidation of uronic acids with Au/Al2O3 catalyst is a highly efficient method to prepare aldaric acids. At pH 8–10 with oxygen bubbling, glucarate and galactarate were quantitatively produced with activities up to 300 mmol per gAu per min and TOF values up to 8000 h−1 which is significant improvement to the methods previously reported. Our results show that alkaline conditions are crucial to the oxidation; pH affects both the initial activity as well as desorption of acids from the catalyst surface. At pH 10, the apparent activation energy Ea for glucuronic acid oxidation was 24.6 kJ mol−1, while at pH 8 Ea increased to 45.3 kJ mol−1 due to the adsorption of free acids on the catalyst surface. Comparison of uronic acid oxidation to their corresponding aldoses, glucose and galactose, indicated that both the stereochemistry of C4 and the carboxylic acid group at C6 affect the oxidation rate. Our observations support the oxidation mechanism, dehydrogenation of an open chain hydrate, proposed for carbohydrate oxidation with Au catalysts. Even though the stability of the Au/Al2O3 catalyst requires further development to prevent Au particle sintering, the selective oxidation of uronic acids is a promising method for production of bio-based chemicals.Acknowledgements
This work was funded by Tekes – the Finnish Funding Agency for Technology and Innovation (project no. 40224/11). The authors would like to thank Electron Microscopy Unit of the Institute of Biotechnology, University of Helsinki for providing laboratory facilities.Notes and references
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- P. Gallezot, Catal. Today, 2007, 121, 76–91 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, U. S. Department of Energy, Golden, CO, 2004, vol. I Search PubMed.
- D. E. Kiely, L. Chen and T. H. Lin, J. Am. Chem. Soc., 1994, 116, 571–578 CrossRef CAS.
- S. Styron, D. Kiely and G. Ponder, J. Carbohydr. Chem., 2003, 22, 123–142 CrossRef CAS PubMed.
- H. Pohjanlehto, H. Setälä, K. Kammiovirta and A. Harlin, Carbohydr. Res., 2011, 346, 2736–2745 CAS.
- C. L. Mehltretter and C. E. Rist, J. Agric. Food Chem., 1953, 1, 779–783 CrossRef CAS.
- D. E. Kiely and K. R. Hash, US Pat., US20080033205, 2008.
- T. N. Smith, K. Hash, C.-L. Davey, H. Mills, H. Williams and D. E. Kiely, Carbohydr. Res., 2012, 350, 6–13 CrossRef CAS PubMed.
- B. Sims, Rivertop contracts with DTI on bioglucarate-based production, 2013, http://biomassmagazine.com/articles/7687/rivertop-contracts-with-dti-on-bioglucarate-based-production/?ref=brm Search PubMed.
- J.-F. Thaburet, N. Merbouh, M. Ibert, F. Marsais and G. Queguiner, Carbohydr. Res., 2001, 330, 21–29 CrossRef CAS.
- M. Ibert, F. Marsais, N. Merbouh and C. Brückner, Carbohydr. Res., 2002, 337, 1059–1063 CrossRef CAS.
- G.-J. t. Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636–1639 CrossRef.
- J. M. H. Dirkx, H. S. van der Baan and J. M. A. J. J. van den Broen, Carbohydr. Res., 1977, 59, 63–72 CrossRef CAS.
- F. R. Venema, J. A. Peters and H. van Bekkum, J. Mol. Catal., 1992, 77, 75–85 CrossRef CAS.
- A. Mirescu and U. Prüße, Appl. Catal., B, 2007, 70, 644–652 CrossRef CAS PubMed.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247 CrossRef CAS.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS PubMed.
- R. Saliger, N. Decker and U. Prüße, Appl. Catal., B, 2011, 102, 584–589 CrossRef CAS PubMed.
- N. Thielecke, M. Aytemir and U. Prüsse, Catal. Today, 2007, 121, 115–120 CrossRef CAS PubMed.
- N. Dimitratos, F. Porta, L. Prati and A. Villa, Catal. Lett., 2005, 99, 181–185 CrossRef CAS PubMed.
- F. M. Gírio, C. Fonseca, F. Carvalheiro, L. C. Duarte, S. Marques and R. Bogel-Łukasik, Bioresour. Technol., 2010, 101, 4775–4800 CrossRef PubMed.
- M. Naghshineh, K. Olsen and C. A. Georgiou, Food Chem., 2013, 136, 472–478 CrossRef CAS PubMed.
- A. Haug and B. Larsen, Acta Chem. Scand., 1963, 17, 1653–1662 CrossRef CAS PubMed.
- K. Rangappa, N. Anitha, M. Nikath, K. Rai and N. Gowda, Synth. React. Inorg. Met.-Org. Chem., 2001, 31, 713–723 CrossRef CAS PubMed.
- V. Shashikala and K. S. Rangappa, J. Carbohydr. Chem., 2002, 21, 491–499 CrossRef CAS PubMed.
- D. Mojzita, M. Wiebe, S. Hilditch, H. Boer, M. Penttilä and P. Richard, Appl. Environ. Microbiol., 2010, 76, 169–175 CrossRef CAS PubMed.
- S. Rautiainen, J. Chen, K. Niemelä, M. Leskelä and T. Repo, 4th Nordic Wood Biorefinery Conference, Helsinki, Finland, 2012, http://www.vtt.fi/inf/pdf/technology/2012/T53.pdf Search PubMed.
- O. A. Simakova, B. T. Kusema, B. C. Campo, A.-R. Leino, K. Kordás, V. Pitchon, P. Mäki-Arvela and D. Y. Murzin, J. Phys. Chem. C, 2011, 115, 1036–1043 CAS.
- B. T. Kusema, B. C. Campo, O. A. Simakova, A.-R. Leino, K. Kordás, P. Mäki-Arvela, T. Salmi and D. Y. Murzin, ChemCatChem, 2011, 3, 1789–1798 CrossRef CAS.
- F. van der Klis, A. E. Frissen, J. van Haveren and D. S. van Es, ChemSusChem, 2013, 6, 1640–1645 CrossRef CAS PubMed.
- S. Ivanova, V. Pitchon, Y. Zimmermann and C. Petit, Appl. Catal., A, 2006, 298, 57–64 CrossRef CAS PubMed.
- I. V. Delidovich, B. L. Moroz, O. P. Taran, N. V. Gromov, P. A. Pyrjaev, I. P. Prosvirin, V. I. Bukhtiyarov and V. N. Parmon, Chem. Eng. J., 2013, 223, 921–931 CrossRef CAS PubMed.
- M. Borrega, K. Niemelä and H. Sixta, Holzforschung, 2013, 67, 871–879 CrossRef CAS PubMed.
- K. Niemelä and E. Sjöström, Carbohydr. Res., 1985, 144, 93–99 CrossRef.
- A. Mirescu, H. Berndt, A. Martin and U. Prüße, Appl. Catal., A, 2007, 317, 204–209 CrossRef CAS PubMed.
- I. R. Siddiqui and C. B. Purves, Can. J. Chem., 1963, 41, 382–386 CrossRef CAS.
- Y. Önal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122–133 CrossRef PubMed.
- A. M. Vannice, Kinetics of Catalytic Reactions, Springer, USA, 2005 Search PubMed.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS PubMed.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1–7 CrossRef CAS PubMed.
- B. T. Kusema, B. C. Campo, P. Mäki-Arvela, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2010, 386, 101–108 CrossRef CAS PubMed.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313–316 CrossRef CAS.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- G. de Wit, J. J. de Vlieger, A. C. Kock-van Dalen, R. Heus, R. Laroy, A. J. van Hengstum, A. P. G. Kieboom and H. van Bekkum, Carbohydr. Res., 1981, 91, 125–138 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst characterization, product analysis, activation energy determination. See DOI: 10.1039/c5ra01802a |
| This journal is © The Royal Society of Chemistry 2015 |