DOI:
10.1039/C5RA01796C
(Paper)
RSC Adv., 2015,
5, 38254-38263
Structure variations of a series of lanthanide complexes constructed from quinoline carboxylate ligands: photoluminescent properties and PMMA matrix doping†
Received
29th January 2015
, Accepted 7th April 2015
First published on 8th April 2015
Abstract
A series of lanthanide complexes with formulae {[KEu(Hqlc)(qlc)(H2O)6(OH)]2+·2Cl−}n (1·Eu), {[Eu(qlc)2(phen)(H2O)2]+·Cl−}·CH3CN (2·Eu), [Eu(qlc)2(phen)(NO3)]·H2O (3·Eu), [Ln(qlc)2(H2O)4]·(qlc)·(H2O) (Ln = Eu(4·Eu), Sm(5·Sm), Gd(6·Gd), Tb(7·Tb), Dy(8·Dy), Ho(9·Ho)) (Hqlc = quinoline-3-carboxylic acid, phen = 1,10-phenanthrolin) are synthesized under solvo(hydro)thermal conditions and characterized by single-crystal X-ray diffraction, infrared spectra, elemental analysis, and powder X-ray diffraction. Complex 1 exhibits two-dimensional (6,3)-connected hcb networks and possesses a stable structure through typical O/C–H⋯Cl intermolecular hydrogen bonds. Complexes 2–4 display three diverse dimer structures, due to the synergistic effect from coordination modes of Hqlc ligand and anion effect. Complexes 5–9 are isostructural with complex 4. Eu-complexes 1–4 could provide intense and bright characteristic 5D0 → 7FJ red luminescence under UV excitation in the solid state at 298 K and 77 K. In complexes 2 and 3, the coordinate phen ligand could play the antenna role in the energy transfer process. Therefore, the luminescence lifetimes of complexes 2 (779.62 and 792.65 μs) and 3 (947.21 and 1095.59 μs) are longer than those of complexes 1 (456.93 and 499.33 μs) and 4 (283.70 and 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 469 μs) in the solid state at 298 K and 77 K. Complexes 5, 7 and 8 exhibit characteristic Sm3+, Tb3+ and Dy3+ ion luminescence. Furthermore, through controlling the concentration of complexes 3 and 4 in poly(methyl methacrylate) (PMMA), a series of 3–PMMA and 4–PMMA hybrid materials are obtained, respectively. They all display strong and characteristic red luminescence emissions at a concentration of 8%. Compared with 3 and 4, the luminescence intensities and luminescence lifetimes of 3–PMMA and 4–PMMA are increased, due to the replacement of water molecules by PMMA.
469 μs) in the solid state at 298 K and 77 K. Complexes 5, 7 and 8 exhibit characteristic Sm3+, Tb3+ and Dy3+ ion luminescence. Furthermore, through controlling the concentration of complexes 3 and 4 in poly(methyl methacrylate) (PMMA), a series of 3–PMMA and 4–PMMA hybrid materials are obtained, respectively. They all display strong and characteristic red luminescence emissions at a concentration of 8%. Compared with 3 and 4, the luminescence intensities and luminescence lifetimes of 3–PMMA and 4–PMMA are increased, due to the replacement of water molecules by PMMA.
Introduction
The versatile photophysical properties of Ln3+ ion include its sharp, intense, and recognizable f → f transitions that are almost independent of the nature of the chemical environment and their excited-state lifetimes, which are usually long enough to allow time-resolved detection. Luminescent lanthanide materials have received increasing attention for applications in the field of smart and hybrid materials.1–4 Whereas, lanthanide ions usually suffer from weak light absorption due to the forbidden f → f transitions (Laporte forbidden), leading to inefficient and inappropriate direct excitation of metal ions.5,6 Through an “antenna effect”, sensitization of the Ln3+ ions with the organic antenna molecule can be achieved by a ligand-to-metal energy-transfer.7–9 To be effective, it is necessary for the energy levels of the donor to be near enough for the acceptor to allow energy transfer occur. Furthermore, the energy levels of the donor must not be located too close so as to result in loss of energy via back-transfer, or other energy migration pathways.10 Among the present sensitizers, several quinoline-based chromophores have attracted attention for constructing luminescent complexes; for example, tri-8-(hydroxyquinoline) aluminum has been developed as an efficient electroluminescence material for organic light emitting diode (OLED) fabrication.11 Besides, various non-covalent weak interactions such as π–π stacking interactions and hydrogen bonding play a significant role to stabilize the supramolecular networks.12–14 Due to the disadvantage of poor mechanical properties of Eu3+ complexes, they are usually applied as organic–inorganic hybrid materials through physical doping or grafting into stable organic or inorganic matrices.15–18 Therefore, when Eu3+ complexes are doped into PMMA to form polymer films, improved flexibility, versatility, and photo-stability properties of luminescent materials could be expected.
Taking account of the above, we prepared a novel series of lanthanide complexes {[KEu(Hqlc)(qlc)(H2O)6(OH)]2+·2Cl−}n (1·Eu), {[Eu(qlc)2(phen)(H2O)2]+·Cl−}·CH3CN (2·Eu), [Eu(qlc)2(phen)(NO3)]·H2O (3·Eu), [Ln(qlc)2(H2O)4]·(qlc)·(H2O) (Ln = Eu(4·Eu), Sm(5·Sm), Gd(6·Gd), Tb(7·Tb), Dy(8·Dy), Ho(9·Ho)) under solvo(hydro)thermal conditions. Quinoline-3-carboxylic acid which comes from quinoline-2,3-carboxylic acid under in situ decarboxylation was studied as a candidate.19 The structure diversities mainly depend on the coordination modes of the Hqlc ligand and counterions. The neutral phen ligand plays the antenna role in the energy transfer process, which enhances the luminescence intensities and luminescence lifetimes of 2 and 3. The photoluminescence properties of 3–PMMA and 4–PMMA are discussed in detail.
Results and discussion
Synthesis and characterization
The reaction routes of 1–4 are shown in Scheme 1. Solvo(hydro)thermal methods20–22 were employed in this work for the synthesis of the complexes. In our work, in situ decarboxylation occurred at quinoline-2,3-dicarboxylic acid (H2qldc) during reaction with lanthanide salts. The ligand 2,3-H2qldc easily lost one 2-position carboxyl group under high temperature (120–160 °C) and produced quinoline-3-carboxylic acid (Hqlc), as shown in Scheme S1.† This result is confirmed by 13C NMR analysis. As shown in Fig. S1,† the characteristic peaks of –COOH appeared at 167.99 and 166.22 ppm for H2qldc, and the reaction product displayed one characteristic peak of –COOH at 166.30 ppm.
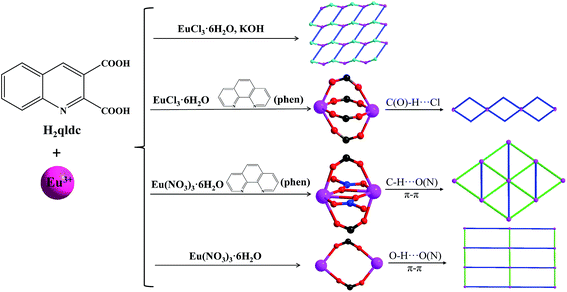 |
| | Scheme 1 The reaction routes of complexes 1–4. | |
In the structure of 1, K+ ions act as structure-directing agents incorporated into the resulting crystalline materials. When the chelate ligand phen was introduced during the assembly of lanthanide complexes to increase the rigidity of the system, dimer complexes 2 and 3 were obtained with different counterions (Cl− and NO3−). Complexes 4–9 are isostructures which are connected by two III–qlc− ligands. Scheme S2† summarizes the coordination modes of the qlc− ligand in the nine complexes. The Cl− ion flexibly coordinates with metal ions or frees in crystal structure to balance the charge (complexes 1 and 2). The Cl− ion is also involved in the construction supramolecules with typical intermolecular hydrogen bonding (O/C–H⋯Cl).23 As for the nitrate anion, it often inclines to strongly chelate to metal ions, acting as a terminal ligand (complex 3 and 4).24 Based on the above analysis, the formation of final complexes are the result of a synergistic effect from coordination modes and the anion effect.
Complexes 1–9 possess the same coordinated qlc− ligand and the IR spectra are similar. The strong and broad absorption bands in the range of 3000–3500 cm−1 in complexes 1–9 are assigned as characteristic peaks of O–H vibration. The strong vibrations appearing around 1600 and 1400 cm−1 correspond to the asymmetric and symmetric stretching vibrations of the carboxylate group, respectively. The absence of strong absorption bands at ca. 1700 cm−1 indicates that the H2qldc ligand is completely deprotonated, as shown in Fig. S2 and S3.† The O–C–O vibration in plane occurs in middle intensity peaks in the range of 580–800 cm−1. Moreover, the weak absorption bands in the IR spectra of 2 and 3 at 464 and 474 cm−1 are assigned to the stretching vibration of the Eu–N bond (Table 1).25
Table 1 Crystal data and structure refinements for complexes 1–9
| |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
| R1 = ∑‖Fo| − |Fc‖/∑|Fo|. wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. |
| Empirical formula |
C20H26N2O11ClEuK |
C34H27ClN5O6Eu |
C32H22N5O8Eu |
C30H28N3O11Eu |
C30H28N3O11Sm |
C30H28N3O11Gd |
C30H28N3O11Tb |
C30H28N3O11Dy |
C30H28N3O11Ho |
| Formula weight |
732.39 |
789.02 |
756.51 |
758.51 |
756.91 |
763.80 |
765.48 |
769.05 |
771.48 |
| Crystal system |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
Pc |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P |
P |
P |
P |
P |
P |
| a (Å) |
7.671(5) |
11.1063(4) |
14.8782(13) |
7.842(5) |
7.8460(16) |
7.8374(16) |
7.834(3) |
7.8379(16) |
7.821(3) |
| b (Å) |
6.820(5) |
11.3995(5) |
13.0813(9) |
11.416(7) |
11.390(2) |
11.382 (2) |
11.385(4) |
11.390(2) |
11.361(3) |
| c (Å) |
25.658(18) |
14.9133(6) |
17.2576(14) |
16.613(10) |
16.620(3) |
16.602(3) |
16.574(6) |
16.582(3) |
16.558(5) |
| α (°) |
90 |
111.9660(10) |
90 |
81.716(7) |
81.81(3) |
81.96(3) |
81.799(4) |
81.94(3) |
81.889(4) |
| β (°) |
94.011(10) |
91.3200(10) |
110.253(2) |
87.825(7) |
87.79(3) |
87.88(3) |
87.851(4) |
87.87(3) |
88.034(12) |
| γ (°) |
90 |
111.1270(10) |
90 |
79.381(7) |
79.34(3) |
79.49(3) |
79.435(4) |
79.35(3) |
79.629(3) |
| Volume (Å3) |
1339.2(16) |
1605.93(11) |
3151.1(4) |
1446.5(15) |
1444.6(5) |
1441.7(5) |
1438.2(9) |
1440.4(5) |
1432.7(8) |
| Z |
2 |
2 |
4 |
2 |
2 |
2 |
2 |
2 |
2 |
| Dcalcd (mg m−3) |
1.186 |
1.632 |
1.595 |
1.742 |
1.740 |
1.759 |
1.768 |
1.773 |
1.788 |
| μ (mm−1) |
2.755 |
2.090 |
2.049 |
2.237 |
2.101 |
2.369 |
2.528 |
2.663 |
2.831 |
| F(000) |
728 |
788 |
1504 |
760 |
758 |
762 |
764 |
766 |
768 |
| θ range (°) |
1.59–24.84 |
3.00–25.76 |
3.11–25.40 |
1.83–25.02 |
3.07–27.48 |
3.08–27.47 |
1.24–26.95 |
3.07–27.48 |
1.24–26.90 |
| Limiting indices |
−9 ≤ h ≤ 9 |
−13 ≤ h ≤ 13 |
−17 ≤ h ≤ 17 |
−9 ≤ h ≤ 9 |
−10 ≤ h ≤ 9 |
−10 ≤ h ≤ 9 |
−9 ≤ h ≤ 9 |
−10 ≤ h ≤ 8 |
−9 ≤ h ≤ 9 |
| 0 ≤ k ≤ 8 |
−11 ≤ k ≤ 13 |
−15 ≤ k ≤ 14 |
−13 ≤ k ≤ 13 |
−14 ≤ k ≤ 14 |
−14 ≤ k ≤ 14 |
−14 ≤ k ≤ 13 |
−14 ≤ k ≤ 14 |
−14 ≤ k ≤ 13 |
| 0 ≤ l ≤ 30 |
−18 ≤ l ≤ 18 |
−20 ≤ l ≤ 20 |
−19 ≤ l ≤ 19 |
−21 ≤ l ≤ 21 |
−21 ≤ l ≤ 21 |
−19 ≤ l ≤ 21 |
−21 ≤ l ≤ 21 |
−20 ≤ l ≤ 19 |
| GOF on F2 |
1.089 |
1.072 |
1.042 |
1.192 |
1.074 |
1.095 |
1.214 |
1.092 |
1.165 |
| Final R indices [I > 2σ(I)] |
R1a |
0.0883 |
0.0282 |
0.0547 |
0.0586 |
0.0353 |
0.0297 |
0.1029 |
0.0361 |
0.0401 |
| wR2b |
0.1973 |
0.0657 |
0.1378 |
0.1645 |
0.0838 |
0.1001 |
0.2206 |
0.0920 |
0.1166 |
| R indices (all data) |
R1 |
0.1181 |
0.0363 |
0.1072 |
0.0640 |
0.0449 |
0.0331 |
0.1074 |
0.0439 |
0.0500 |
| wR2 |
0.2189 |
0.0757 |
0.1690 |
0.1667 |
0.0875 |
0.1075 |
0.2228 |
0.0960 |
0.1257 |
Crystal structures
{[KEu(Hqlc)(qlc)2(H2O)6(OH)]2+·2Cl−}n (1). Single-crystal X-ray diffraction analysis reveals that complex 1 crystalizes in the monoclinic Pc space group, which exhibits a 2D (6,3)-connected hcb network. The asymmetric unit of 1 consists of one crystallographically unique Eu3+ cation, one K+ cation, one fully-deprotonated qlc− anion, one Hqlc ligand, six coordinated water molecules, one OH− anion, and two free Cl− anions. Each central Eu3+ ion is nine-coordinated by two oxygen atoms (O1 and O4) from two different Hqlc ligands, seven oxygen atoms from six water molecules and OH− anions resulting in the tricapped trigonal prism geometry, as illustrated in Fig. S4.† The Eu–O distances vary from 2.282(17) to 2.603(11) Å. Each K+ ion is four-coordinated by three oxygen atoms from three different water molecules and one nitrogen atom (N1) from II–Hqlc ligand. Thus, the four-coordinated K+ atom forms a tetrahedron. The K–O distances are 3.049(16), 3.060(15), and 3.202(19) Å, respectively, and K–N distance is 3.030(20) Å, which are in accordance with reported values in other europium–potassium complexes.26 Adjacent two Eu3+ ions linked two I–qlc− ligands at both sides that are bridged by potassium and water molecules to form an infinite 1D chain structure (Fig. 1a). The 1D chains are further bridged through the N1 atom from II–Hqlc ligand forming a 2D layer structure (Fig. 1b). If you ignore the K+ ion, complex 1 is only a discrete structure. Therefore, the K+ ion directs and changes the structure formation of 1. Furthermore, the H⋯Cl distances between the OH and chlorine atoms from neighboring chains are shorter than the sum of the van der Waals radii for H and Cl (ca. 1.2 Å for H, 1.75 Å for Cl), and the angles are in the range of 131.22–163.93°, which indicates that the typical intermolecular hydrogen bonds existence in the structure of 1, as shown in Fig. 1c. If we ignore the I–qlc− ligand, the metal centers (Eu and K atoms) are viewed as the nodes and the Hqlc ligands as linkers, the resulting 2D structure may be simplified into a (6,3)-connected hcb network as depicted in Fig. 1d.
 |
| | Fig. 1 (a) Ball-and-stick representation of the 1D chain structure. (b) The 2D layer structure of 1. (c) View of the intermolecular hydrogen bonding O–H⋯Cl in complex 1 (hydrogen bonds are shown by the green dashed lines). (d) The 2D (6,3)-connected hcb topology in its symmetrical form is distinguished by different colors (Eu3+, pink ball; K+, light blue ball; Hqlc, deep blue linkers). | |
{[Eu(qlc)2(phen)(H2O)2]+·Cl−}·CH3CN (2). Single-crystal X-ray diffraction analysis reveals that complex 2 crystalizes in the triclinic P space group, which features a dimer structure connected by a Hqlc ligand. The asymmetric unit of 2 consists of one crystallographically unique Eu3+ cation, two fully-deprotonated qlc− anions, one phen, two coordinated water molecules, one free CH3CN and free Cl− anion, as shown in Fig. 2a. Each central Eu3+ ion is eight-coordinated by four oxygen atoms (O1, O2A, O3, and O4) from four qlc− ligands, two oxygen atoms (O5 and O6) from two H2O molecules, and two nitrogen atoms (N1 and N2) from a phen ligand, resulting in the bicapped trigonal prism geometry (Fig. S5†). The distances between the two coordinated nitrogen atoms and Eu3+ are 2.594 (8) Å (Eu1–N1) and 2.647 (8) Å (Eu1–N2). The Eu–O distances vary from 2.319 (3) to 2.480 (3) Å in accordance with reported values in other europium complexes.27 The average bond length of the Eu–Ocarboxyl is 2.341 Å, which is slightly shorter than that of Eu–Owater (2.452 Å). This is attributed to the fact that Eu–Ocarboxyl is stronger than the covalent bond of Eu–Owater. Eu1 and its corresponding centrosymmetric generated atom Eu1A (symmetry code A = 2 − x, 2y, 1 − z) are joined by two bidentate bridged carboxylic groups (III–qlc−) to form a binuclear unit, with the separation of Eu⋯Eu being 4.364 Å, as shown in Fig. 2b. The cationic charge [Eu(qlc)2(phen)(H2O)2]+ moieties is balanced by the one free Cl− anion, which can serve as the hydrogen bond acceptor. The 1D chain structure is obtained by intermolecular hydrogen bonding interactions which are formed by uncoordinated Cl− anions and coordinated water molecules (O5 and O6). Furthermore, the C25–H25⋯Cl1 hydrogen bonding interaction is involved in the structure stability (Fig. 2c). If considering the dimer structure as nodes and these hydrogen bonds (O–H⋯Cl and C–H⋯Cl) as ‘V’-shaped linkers, a beaded chain of 2 is created (Fig. 2d).
 |
| | Fig. 2 (a) The metal coordination environment in 2 with labeling scheme and 50% thermal ellipsoids (hydrogen atoms and free CH3CN are omitted for clarity). (b) Ball-and-stick representation of the dimer structure of complex 2. (c) 1D supramolecular structure of 2 (hydrogen bonding is shown by the green dashed lines). (d) The beaded chain structure of complex 2. | |
[Eu(qlc)2(phen)(NO3)]·H2O (3). Single-crystal X-ray diffraction analysis reveals that complex 3 crystalizes in the monoclinic P21/n space group, which features a dimer structure connected by NO3− and qlc− ligand. As shown in Fig. S6,† the asymmetric unit of 3 consists of one crystallographically unique Eu3+ cation, two fully-deprotonated qlc− anions, one phen ligand, one nitrate anion, and one free water molecule. Each Eu3+ center is nine-coordinated with seven oxygen atoms, including four carboxylic oxygen atoms (O1, O2A, O3, and O4) from three qlc− ligands, and three oxygen atoms (O5, O5A, and O6) from two NO3− anions. The uncoordinated sites are occupied by two nitrogen atoms (N3 and N4) from the phen ligand. As a result, the coordination geometry around the metal center can be best described as a distorted tricapped trigonal prism geometry. Eu1 and its corresponding centrosymmetric generated atom Eu1A (symmetry code A = 1 − x, 2y, −z) are joined by two bidentate bridged carboxylic groups (III–qlc−) and NO3− anions to form a binuclear unit, with the separation of Eu⋯Eu being 3.943 Å (Fig. 3a). This distance is the shortest among complexes 2–4, because the NO3− anions connected to the adjacent metal centers narrows the distance between the two metal centers. It is worth noting that π⋯π interactions and hydrogen bonding play critical roles to stabilize the structure.28 The pyridyl ring of qlc− ligand and phen plane take place in a face-to face mode, with the intercentroid distances 3.589 Å (Fig. 3b). The moieties are extended into a 2D supramolecular framework through the C–H⋯N and C–H⋯O hydrogen bonding interactions (C22–H22A⋯N2 = 2.661 Å and C25–H25A⋯O6 = 2.608 Å), as displayed in Fig. 3c. If the dimer structure is viewed as the node, and hydrogen bonds or π⋯π stacking interactions as linkers, the resulting 2D supramolecular structure may be simplified into a “dense” (3,6) network, as shown in Fig. 3d.
 |
| | Fig. 3 (a) Ball-and-stick representation of the dimer structure of complex 3. (b) The π–π interactions between the two face-to-face pyridyl rings and benzene ring. (c) A 2D supramolecular structure of complex 3 constructed by hydrogen bonds and π⋯π stacking interactions along the a axis. (d) View of the 2D supramolecular layer (3,6) network in complex 3. | |
[Ln(qlc)2(H2O)4]·(qlc)·(H2O) (Ln = Eu(4), Sm(5), Gd(6), Tb(7), Dy(8), Ho(9)). Single-crystal X-ray diffraction analysis reveals that complexes 4–9 are isomorphous and crystallize in the triclinic P space group, which features a dimer structure. Only the structure of 4 is described in detail. The asymmetric unit of 4 consists of one independent Eu3+ cation, two fully-deprotonated qlc− anions, four coordinated water molecules, one free fully-deprotonated qlc− anion and one free water molecule all in general positions. The Eu3+ center is eight coordinated, with four oxygen atoms (O1A, O2, O3, and O4) derived from two qlc− ligands and four oxygen atoms (O7, O8, O9, and O10) derived from four terminal coordinated water molecules, forming a distorted bicapped trigonal prism geometry, as displayed in Fig. S7.† The bond lengths of Eu–O vary from 2.303 (7) to 2.453 (8) Å. O–Eu–O band angles are in the range of 53.2 (3)–154.0 (3)°. All the Eu–O distances and O–Eu–O bond angles are close to the values found in other Eu3+ complexes.29 The average bond length of Ln–O(carboxyl) decreases with decreasing radii of the Ln ions.30 Two equivalent Eu3+ centers are bridged by the III–qlc− ligand to give a dimer lanthanide structure with separations of Eu⋯Eu (5.320 Å), as shown in Fig. 4a. The Eu⋯Eu distance is the longest among the four complexes as the adjacent metal centers are connected through two III–qlc− ligands, the carboxylic acid group can stretch better. In complex 4, the following ones can be found: O–H⋯O, O–H⋯N, donor⋯acceptor distance is in the range of 2.723–3.354 Å. The shortest distance 2.723 Å refers to the intramolecular O7–H4C⋯O6 (∠OHO = 117.73°) hydrogen bond. The intermolecular hydrogen bonds connected the dimer structure into 1D chains (Fig. 4b). The 1D chains are further assembled into 2D supramolecular layer structures through inter-layer π–π stacking interactions between the adjacent quinolone rings, as shown in Fig. 4c. The π–π stacking distance between two benzene rings is 3.351 Å. The two quinoline-ring planes have nearly parallel geometry with the dihedral angle at 1.017°. Similar to complexes 2 and 3, through these weak interactions a 2D supramolecular network is constructed, as shown in Fig. 4d, in which each dimer links four adjacent ones. Thus, the overall topology of 4 can be defined as 44 topology. The hydrogen bonding interactions in complexes 1–9 are listed in Table S3.†
 |
| | Fig. 4 (a) Ball-and-stick representation of the dimer structure of 4. (b) A 1D supramolecular structure constructed by intermolecular hydrogen bonds O–H⋯O and O–H⋯N in complex 4. (c) A 2D supramolecular network constructed by π–π stacking interactions between the two face-to-face pyridyl rings. (d) View of the 2D supramolecular layer structure with 44 topology. | |
Powder X-ray diffraction (PXRD) has been used to check the phase purity of the bulky samples in the solid state. As shown in Fig. S8,† the experimental PXRD patterns of 1–9 are in good agreement with those simulated from their single-crystal X-ray data, indicating the purity of the as-synthesized products. The differences in intensities may be due to the preferred orientation of the powder samples.
Photoluminescence properties
The luminescent properties of the ligands and complexes 1–8 were examined in the solid state at 298 K or 77 K and are summarized in Table S4,† Fig. 5–7 and S10–S12.† Upon excitation from 320 to 365 nm, which are the maximum of the excitation spectra, complexes 1–4 show the characteristic narrow emission bands of the Eu3+ ion corresponding to the 5D0 → 7FJ (J = 0–4) transitions, as illustrated in Fig. 5. Among them, the 5D0 → 7F2 transition at λ = 613 nm is the strongest emission that is an induced electric dipole transition, and its corresponding intensity is very sensitive to the coordination environment. The very intense 5D0 → 7F2 peak, points to a highly polarizable chemical environment around the Eu3+ ion and is responsible for the brilliant red emission of complexes 1–4. Furthermore, the emission spectra of complexes 1–4 show only one peak for the 5D0 → 7F0 transition and three stark components for the 5D0 → 7F1 transition, indicating the presence of a single chemical environment around the Eu3+ ion. The emission bands around 578 and 650 nm are very weak, since their corresponding transitions 5D0 → 7F0,3 are forbidden both in the magnetic and electric dipole schemes. The emission band at 699 nm is attributed to 5D0 → 7F4 transition. The absence of bands from higher excited states such as 5D1 in the emission spectrum implies that the nonradiative relaxation to the 5D0 level is efficient.31 Additionally, the luminescent intensities of 1–4 in the solid state at 77 K are higher than those at 298 K, due to the O–H oscillators being protected at low temperature.5
 |
| | Fig. 5 Solid-state excitation and emission spectra of complexes 1–4 both at 298 K and 77 K (the inset shows the photographs of complex 3 under a UV lamp). | |
 |
| | Fig. 6 Excitation and emission spectra of complex 7 in the solid state at 298 K and 77 K. | |
 |
| | Fig. 7 Emission spectra of complexes 5 (left) and 8 (right) in the solid state at 298 K and 77 K. | |
The decay curves of 1–4 were measured at both 298 K and 77 K and monitored within the most intense lines of the 5D0 → 7F2 transition. The observed luminescent decay profiles correspond to single exponential functions at 298 K and 77 K. The luminescence lifetimes in the solid state are determined to be 456.93, 779.62, 947.21, and 283.70 μs at 298 K. At 77 K, the τ values (499.33 for 1, 792.65 for 2, 1098.59 for 3, and 464.69 μs for 4) are much higher than those at 298 K to 77 K, due to the absence of thermally activated deactivation processes. These complexes show the increasing luminescence lifetime as a sequence 4 < 1 < 2 < 3 both at 298 K and 77 K. The shortest 5D0 lifetime noted for the Eu3+ complex 4 may be due to the dominant nonradiative decay channels associated with vibronic coupling because of the presence of four coordinate water molecules. As a result of the potassium-to-europium charge transfer (MMCT)32 in 1, the luminescence lifetime of 1 is longer than 4, though there are more water molecules in 1. The photoluminescence is closely related to the local environments around metal ions, as the Eu⋯Eu distances and Eu–N bond lengths are shortest in complex 3, the energy levels are lowered. Therefore, the luminescence lifetime of 3 is the longest.
As shown in Fig. 6, the excitation spectra of complex 7, which monitored the intense characteristic emission (545 nm) of the Tb3+ ion, displays a broad band with a peak at 360 (298 K) and 330 nm (77 K) for the electronic transitions of Hqlc ligand. The emission spectrum of 7 shows characteristic Tb3+ metal-centered and ligand-centered luminescence, respectively. The ligand-centered emission (412 nm at 298 K and 423 nm at 77 K) is red-shifted compared with the free ligand (400 nm), which may be attributed to the metal-disturbed ligand-centered π* → π transitions.33 Complex 7 shows four line-like emission bands at 490, 545, 584, and 621 nm, which results from deactivation of the 5D4 excited state to the corresponding ground states 7FJ (J = 6, 5, 4, 3) of Tb3+ ion. The 5D4 → 7F5 transition at 545 nm is the strongest emission, implying intense green luminescence. Besides, the transitions to 7FJ (J = 2, 1, 0) levels are too weak to measure.34 The luminescence lifetime of complex 7 at 77 K (8.77 μs) is longer than at 298 K (5.55 μs).
The solid state visible luminescence of complexes 5 and 8 were investigated at room temperature and liquid nitrogen temperature (Fig. 7). Upon excitation at 325 nm, which is the maximum of the excitation spectrum, complex 5 shows characteristic narrow band emissions of the Sm3+ ion corresponding to the 4G5/2 → 6H5/2 (562 nm), 4G5/2 → 6H7/2 (596 nm), 4G5/2 → 6H9/2 (644 nm), and 4G5/2 → 6H11/2 (700 nm), as shown in Fig. 8. The ligand-centered emission (430 nm at 298 K and 415 at 77 K) is red-shifted compared with the free ligand, which may be attributed to the metal disturbed ligand-centered π* → π transitions.33 The lifetimes of 4G5/2 amounts to 21.43 μs at 298 K and 21.77 μs at 77 K. Upon excitation at 325 nm, complex 8 shows characteristic narrow band emissions of Dy3+ ion corresponding to 4F9/2 → 6H15/2 (481 nm), 4F9/2 → 6H13/2 (574 nm), 4F9/2 → 6H11/2 (661 nm), and 4F9/2 → 6H9/2 + 6F11/2 (750 nm). The characteristic 4F9/2 → 6H9/2 + 6F11/2 transition is rarely observed and described.35 We have observed ligand-centered π* → π transitions (420 nm at 298 K and 405 nm at 77 K). The lifetimes of 4F9/2 are 7.09 μs at 298 K and 7.97 μs at 77 K.
 |
| | Fig. 8 Excitation and emission spectra of 3–PMMA and 4–PMMA films doped with 2–10% of 3 and 4. | |
We have also observed emissions in the near infrared region from the Sm3+ and Dy3+ ions (Fig. S11†), which is a rarely described phenomenon.36,37 The emission spectrum of 5 consists of several bands at λ = 936, 988, 1193, 1285, and 1383 nm. Emission peaks at 936 nm, 988 nm and 1193 nm and 1196 nm are suspected to be the Stark splitting of 6F5/2, 6F7/2 and 6F9/2, respectively. The other emissions are assigned to the f–f transitions of 6F11/2 (1285 nm and 1383 nm), respectively. The emission bands of 5 are shifted relative to the bands of the reported theoretical values. The emission spectrum of 8 consists of several bands at λ = 948, 985, and 1187 nm, which are attributed to the f–f transitions 4F9/2 → 6F7/2, 4F9/2 → 6F5/2, and 4F9/2 → 6F3/2, respectively.
Energy transfer process
The data of the triplet excited state T1 of the ligand (23809 cm−1) was calculated by the low-temperature (77 K) phosphorescence spectrum of complex 6. The singlet energy level (S1) of the Hqlc ligand is 28169 cm−1 (355 nm), which is calculated from the UV-Vis absorbance edges (Fig. S12†). The singlet and triplet energy levels of phen (31000 and 22100 cm−1) were taken from the literature.38 In a typical example of complex 3, the energy transfer process is discussed. As it is known that the intersystem crossing process becomes effective when ΔE (1ππ*–3ππ*) is at least 5000 cm−1, the energy gap ΔE (1ππ*–3ππ*) for Hqlc ligand and phen are 4369 cm−1 and 8900 cm−1, respectively. This indicates that the intersystem crossing is effective in the phen ligand.39 Otherwise, the energy gap between 3ππ* (qlc− ligand) and 1ππ* (phen ligand) is 6069 cm−1. This indicates that the energy may transfer from qlc− to the phen ligand. The energy difference between the lowest triplet state of phen and the resonant energy levels of Eu3+ (5D1, 18674 cm−1) is 3426 cm−1. According to the empirical rule proposed by Latva,40 an optimal ligand-to-metal energy transfer process for Eu3+ needs 2500–4500 cm−1, the energy differences in Scheme 2 therefore show that the transition is effective from the triplet energy level of phen to Eu3+ ion. The absence of bands from higher excited states such as 5D1 in the emission spectrum implies that the nonradiative relaxation to the 5D0 level is efficient. According to the consideration of the above, the emission intensity and luminescence lifetime of complexes 2 and 3 are enhanced because the ligand-to-metal energy transfer processes could occur in the mixed ligand complexes.
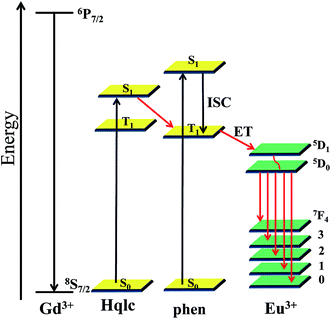 |
| | Scheme 2 Schematic energy level diagram and energy transfer process. | |
Properties of complexes 3 and 4 doped into PMMA polymer films
In consideration of the excellent performance of PMMA as one of the most popular polymer matrices with low cost, easy preparation, and good mechanical properties,41,42 two PMMA-supported hybrid materials 3–PMMA and 4–PMMA are obtained from complexes 3 and 4, respectively. The excitation and emission spectra of the PMMA polymer doped with complexes 3 and 4 at different concentrations (2%, 4%, 6%, 8%, and 10%) is shown in Fig. 8. The excitation spectra are dominated by intense broad bands at 325 nm and 330 nm, which can be assigned to absorptions of both the PMMA polymer and the organic chromophore. The emission spectra 3–PMMA and 4–PMMA at a variety of concentrations exhibits five emission bands that are assigned to the characteristic 5D0 → 7FJ (J = 0–4) transitions of Eu3+ ion. The luminescent intensity of the Eu3+ emission at 613 nm is strengthened as the concentration of complexes 3 and 4 increases and reaches a maximum at a concentration of 8%. Further increases in the concentration lead to a decrease the luminescent intensity. The energy transfer between the lanthanide ions themselves is a nonradiative process, which accounts for the decrease in the Eu3+ emission, especially at a high concentration (10%).43 The luminescence decay curves of the doped films were obtained by monitoring the emission at the hypersensitive 5D0 → 7F2 (613 nm). These data were adjusted with mono-exponential decay function and the lifetime values (τ) of the emitter 5D0 level of the doped systems were determined and are listed in Table S5.† As shown in Fig. S13,† the luminescence lifetimes for 3–PMMA and 4–PMMA are higher than complexes 3 and 4, thus indicating that radiative process are operative in the doped polymer films because of the absence of O–H oscillators from the water molecules. Comparing the emission spectra of 3, 4, 3–PMMA and 4–PMMA, the luminescence intensities are in the sequence of 3–PMMA > 3 > 4–PMMA > 4. The reason for the 3 > 4 emission intensity is due to the substitution of the water molecules by the bidentate nitrogen donors in complex 3. Luminescent lifetime of 4–PMMA (τ = 972.33 μs) increases nearly three times longer than complex 4 (τ = 283.70 μs) and the luminescent intensity is twice that of 4 (Fig. S14†), as the structure of 4 has more coordinate water molecules than 3.
Conclusion
In summary, a series of lanthanide complexes have been synthesized with H2qldc under solvo(hydro)thermal conditions. The K+ ion acts as a structure-directing agent and is connected adjacent to the Eu3+ ion through water molecules and nitrogen atom from Hqlc ligand to form a 2D (6,3)-connected hcb network in 1. Complexes 2–4 display three diverse dimer structures due to the different coordination modes and anion effect. These complexes show increasing luminescence lifetimes in the sequence 4 < 1 < 2 < 3 both at 298 K and 77 K, which are closely related to their structure models. The photoluminescence properties study on complexes 1–4 demonstrates that the Eu3+ luminescence is well-sensitized by Hqlc and phen ligands, indicating an efficient energy transfer process from ligands to Eu3+ ions in complexes 2 and 3. Moreover, PMMA enhances the luminescence intensity and luminescence lifetimes of the PMMA film doped with complexes 3 and 4 in comparison with the precursor complexes. Therefore, the PMMA doped with the luminescent lanthanide complexes may be of potential application as light-emitting materials and active polymer optical devices.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant 21371040 and 21171044), the National Key Basic Research Program of China (973 Program, no. 2013CB632900), the Fundamental Research Funds for the Central Universities (Grant no. HIT. IBRSEM. A. 201409), and the Program for Innovation Research of Science in Harbin Institute of Technology (PIRS of HIT no. A201416 and B201414).
References
- N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dincaã, J. Am. Chem. Soc., 2013, 135, 13326 CrossRef CAS PubMed.
- J. N. Hao and B. Yan, J. Mater. Chem. C, 2014, 2, 6758 RSC.
- Z. Zhang, W. X. Feng, P. Y. Su, X. Q. Lü, J. R. Song, D. D. Fan, W. K. Wong, R. A. Jones and C. Y. Su, Inorg. Chem., 2014, 53, 5950 CrossRef CAS PubMed.
- P. R. Matthes, J. Nitsch, A. Kuzmanski, C. Feldmann, A. Steffen, T. B. Marder and K. Müller-Buschbaum, Chem.–Eur. J., 2013, 19, 17369 CrossRef CAS PubMed.
- H. B. Zhang, L. J. Zhou, J. Wei, Z. H. Li, P. Lin and S. W. Du, J. Mater. Chem., 2012, 22, 21210 RSC.
- Z. Q. Xia, Q. Wei, Q. Yang, C. F. Qiao, S. P. Chen, G. Xie, G. C. Zhang, C. S. Zhou and S. L. Gao, CrystEngComm, 2013, 15, 86 RSC.
- S. Biju, M. L. P. Reddy, A. H. Cowley and K. V. Vasudevan, Cryst. Growth Des., 2009, 9, 3562 CAS.
- M. Hatanaka and K. Morokuma, J. Chem. Theory Comput., 2014, 10, 4184 CrossRef CAS.
- R. Shyni, S. Biju, M. L. P. Reddy, A. H. Cowley and M. Findlater, Inorg. Chem., 2007, 46, 11025 CrossRef CAS PubMed.
- J. Gregolińshi, P. Starynowicz, K. T. Hua, J. L. Lunkley, G. Muller and J. Lisowski, J. Am. Chem. Soc., 2008, 130, 17761 CrossRef PubMed.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS PubMed.
- M. F. Wang, X. J. Hong, Q. G. Zhan, H. G. Jin, Y. T. Liu, Z. P. Zheng, S. H. Xu and Y. P. Cai, Dalton Trans., 2012, 11898 RSC.
- G. Holger and K. Gerhard, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef.
- K. Müeller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143 CrossRef PubMed.
- T. W. Duan and B. Yan, J. Mater. Chem. C, 2014, 2, 5098 RSC.
- L. D. Carlos, R. A. S. Ferreira, V. Z. Bermudez, B. Julián-López and P. Escribano, Chem. Soc. Rev., 2011, 40, 536 RSC.
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696 RSC.
- S. Rodríguez, P. Elizondo, S. Bernès, N. Pérez, R. Bustos and E. García-España, Polyhedron, 2015, 85, 10 CrossRef PubMed.
- C. B. Liu, Q. Li, X. Wang, G. B. Che and X. J. Zhang, Inorg. Chem. Commun., 2014, 39, 56 CrossRef CAS PubMed.
- N. Wei, M. Y. Zhang, X. N. Zhang, G. M. Li, X. D. Zhang and Z. B. Han, Cryst. Growth Des., 2014, 14, 3002 CAS.
- T. J. Sørensen, L. R. Hill, J. A. Tilney, O. A. Blackburn, M. W. Jones, M. Tropiano and S. Faulkner, Eur. J. Inorg. Chem., 2014, 15, 2520 CrossRef PubMed.
- K. Miyata, T. Nakanishi, K. Fushimi and Y. Hasegawa, J. Photochem. Photobiol., A, 2012, 235, 35 CrossRef CAS PubMed.
- C. X. Ding, X. Rui, C. Wang and Y. S. Xie, CrystEngComm, 2014, 16, 1010 RSC.
- X. P. Yang, D. Schipper, L. J. Zhang, K. Q. Yang, S. M. Huang, J. J. Jiang, C. Y. Su and R. A. Jones, Nanoscale, 2014, 6, 10569 RSC.
- A. Q. Zhang, J. L. Zhang, Q. L. Pan, S. H. Wang, H. S. Jia and B. S. Xu, J. Lumin., 2012, 132, 965 CrossRef CAS PubMed.
- J. W. Dai and M. L. Tong, CrystEngComm, 2012, 14, 2124 RSC.
- J. H. Liao, W. S. Hwang and G. Y. Chen, Z. Anorg. Allg. Chem., 2014, 640, 1793 CrossRef CAS PubMed.
- K. M. Sureshan and R. G. Gonnable, CrystEngComm, 2013, 15, 1676 RSC.
- M. Zhu, Z. M. Hao, X. Z. Song, X. Meng, S. N. Zhao, S. Y. Song and H. J. Zhang, Chem. Commun., 2014, 50, 1912 RSC.
- G. L. Zhuang, X. J. Kong, L. S. Long, R. B. Huang and L. S. Zheng, CrystEngComm, 2010, 12, 2691 RSC.
- D. B. A. Raj, S. Biju and M. L. P. Reddy, Inorg. Chem., 2008, 27, 8091 CrossRef PubMed.
- Z. P. Zheng, Y. J. Qu, X. J. Hong, L. M. Wei, L. T. Wan, W. H. Zhou, Q. G. Zhan and Y. P. Cai, Inorg. Chem., 2014, 53, 9625 CrossRef CAS PubMed.
- S. Chen, R. Q. Fan, S. Gao, X. M. Wang and Y. L. Yang, J. Lumin., 2014, 149, 75 CrossRef CAS PubMed.
- J. D. Xu, T. M. Corneillie, E. G. Moore, G. L. Law, N. G. Butlin and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 19900 CrossRef CAS PubMed.
- P. Wang, R. Q. Fan, Y. L. Yang, X. R. Liu, P. Xiao, X. Y. Li, W. L. J. Hasi and W. W. Cao, CrystEngComm, 2013, 15, 4489 RSC.
- B. Chu, W. L. Li, Z. R. Hong, F. X. Zang, H. Z. Wei, D. Y. Wang, M. T. Li, X. C. S. Lee and S. T. Lee, J. Phys. D: Appl. Phys., 2006, 39, 4549 CrossRef CAS.
- S. Q. Su, W. Chen, C. Qin, S. Y. Song, Z. Y. Guo, G. H. Li, X. Z. Song, M. Zhu, S. Wang, Z. M. Hao and H. J. Zhang, Cryst. Growth Des., 2012, 12, 1808 CAS.
- D. B. Ambili Raj, B. Francis, M. L. P. Reddy, R. R. Butorac, V. M. Lynch and H. Cowley, Inorg. Chem., 2010, 49, 9055 CrossRef PubMed.
- R. Shyni, S. Biju, M. L. P. Reddy, A. H. Cowley and M. Findlater, Inorg. Chem., 2007, 46, 11025 CrossRef CAS PubMed.
- W. B. Sun, P. F. Yan, G. M. Li, H. Xu and J. W. Zhang, J. Solid State Chem., 2009, 182, 381 CrossRef CAS PubMed.
- T. R. Wang, P. Li and H. R. Li, ACS Appl. Mater. Interfaces, 2014, 6, 12915 CAS.
- P. Martín-Ramos, V. Lavín, M. R. Silva, I. R. Martín, F. Lahoz, P. Chamorro-Posada, J. A. Paixão and J. Martín-Gil, J. Mater. Chem. C, 2013, 1, 5701 RSC.
- S. Biju, Y. K. Eom, J. G. Bünzli and H. K. Kim, J. Mater. Chem. C, 2013, 1, 6935 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1041890–1041898. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra01796c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 









