
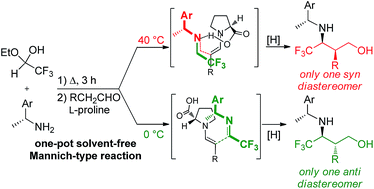
Trifluoromethyl syn- or anti-γ-amino alcohols by one-pot solvent-free Mannich-type reactions under temperature control†
Abstract
Starting from trifluoroacetaldehyde ethyl hemiacetal, chiral amines and suitable aldehydes, diastereomerically pure fluorinated syn- or anti-γ-amino alcohols can be obtained by a friendly one-pot solvent-free L-proline catalysed Mannich-type reaction only by changing the temperature.
 Please wait while we load your content...
Please wait while we load your content...

