
Use of a tin antimony alloy-filled porous carbon nanofiber composite as an anode in sodium-ion batteries
Abstract
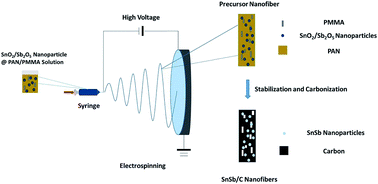
Lithium-ion battery is currently the dominant energy storage technology for electronic devices and electric vehicles. However, the predictable rising cost of lithium raw materials has attracted increasing interest in less expensive rivals, such as sodium-ion battery. In this work, a tin antimony (SnSb) alloy-filled porous carbon nanofiber composite was prepared as a sodium-ion battery anode material by a simple electrospinning method with subsequent thermal treatment. The spinning solution contained antimony tin oxide nanoparticles as the SnSb alloy precursor, polyacrylonitrile as the carbon precursor, and polymethyl methacrylate (PMMA) as the pore generator. The resultant SnSb@C nanofiber composite formed a continuous conductive network, which was favorable for enhancing its electrochemical performance. The presence of the SnSb alloy significantly increased the energy storage capacity of the composite due to its high theoretical capacity. The porous structure created by the decomposition of the PMMA polymer provided a free space to buffer the volume change of the SnSb alloy during the sodiation–desodiation process. The resultant SnSb@C nanofiber composite exhibited high capacity and a stable rate capability, and it was demonstrated to be a promising anode candidate for sodium-ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

