
Characterization of stress degradation products of mirabegron using UPLC-QTOF-MS/MS and in silico toxicity predictions of its degradation products
Pradipbhai D. Kalariyaa,
Mahesh Sharmab,
Prabha Gargb,
Jagadeshwar Reddy Thotad,
Srinivas Ragampetaac and
M. V. N. Kumar Talluri*a
aDepartment of Pharmaceutical Analysis, National Institute of Pharmaceutical Education & Research, IDPL R&D Campus, Balanagar, Hyderabad-500 037, India. E-mail: narendra.talluri@gmail.com; narendra@niperhyd.ac.in; Fax: +91-40-23073751; Tel: +91-40-23423749 ext. 2012
bDepartment of Pharmacoinformatics, National Institute of Pharmaceutical Education and Research (NIPER), Sector-67, S.A.S (Mohali). Nagar, Punjab-160062, India
cNational Center for Mass Spectrometry, CSIR-Indian Institute of Chemical Technology, Tarnaka, Hyderabad 500607, India
dSophisticated Analytical Instrument Facility, CSIR-Central Drug Research Institute, Lucknow-226021, India
First published on 26th March 2015
Abstract
Mirabegron is a novel beta-3 adrenergic receptor agonist in the treatment of overactive bladder disorder. The drug was subjected to hydrolytic, photolytic, thermal and oxidative stress conditions as per the International Conference on Harmonization guidelines (ICH) Q1A (R2) to understand the degradation profile of the drug. The safety of the drug may be affected by degradation products present in the drug. As a result, identification and characterization of degradation products has become very important in drug development processes. In this study, a simple, rapid, precise and accurate ultra performance liquid chromatography (UPLC-PDA) method has been developed on a Waters CSH C18 column (100 mm × 2.1 mm, 1.7 μm) using gradient elution of ammonium acetate (10 mM, pH 5) and acetonitrile as mobile phase. Mirabegron was found to degrade under hydrolytic and oxidative stress conditions while it was stable under thermal and photolytic conditions. A total of seven degradation products were characterized by UPLC-MS/MS in positive ion mode, combined with accurate mass measurements. The proposed structures of the degradation products have been rationalized by appropriate mechanisms. Additionally, in silico toxicity was predicted for all degradant products by using TOPKAT and DEREK softwares to enhance the safety of the drug.
Introduction
Mirabegron (MIR) is being developed as a novel beta-3 adrenergic agonist for the treatment of an overactive bladder (OAB). It is a urological disorder characterized by the bothersome symptoms of urgency, increased voiding frequency or incontinence and nocturia. Currently, the muscarinic receptor antagonists are the frontline treatment for OAB. However, side effects such as blurred vision, dry mouth and occasionally urinary retention are associated with these drugs.1 Hence, a novel target has emerged for treatment of OAB to overcome these side effects. It has been proved that beta-3 adrenoreceptors cause detrusor muscle relaxation in the human bladder. Hence, they are a potential target to treat OAB. Mirabegron binds with beta-3 adrenoreceptors and has been shown to decrease the muscle contractions.2–5 It is chemically known as 2-(2-aminothiazol-4-yl)-N-[4-(2-{[(2R)-2-hydroxy-2-phenylethyl]amino}ethyl) phenyl]acetamide.A thorough literature search revealed that RP-HPLC method for pharmaceutical dosage form,6 GC method for residual solvent analysis7 and one bioanalytical method has been reported for the determination of MIR in the human plasma by LC-MS/MS for clinical pharmacokinetic study.8 To the best of our information, a comprehensive stress degradation study has not been reported so far. Identification of stability affecting factors such as temperature, oxidation, light and pH would facilitate the selection of storage conditions, packaging materials and handling conditions. Hence, regulatory bodies have set stress studies and characterization of degradation products (DPs) as mandatory requirements.9,10 Further characterization of unknown degradation impurities is also required to ascertain that an impurity does not have any genotoxic concern. Nowadays, in silico toxicity methodology has been emerging as a useful tool for prediction of toxicity of the molecules. In 2006, the EU completely revised its regulatory structure for chemicals with the passage of the regulation concerning registration, evaluation, authorization, and restriction of chemicals (REACH). Alternative testing methods are urgently needed to fulfill the goal of reducing animal testing in REACH. The REACH regulation mentions non-testing methods for “predictive toxicology” in risk assessment of the drug and its possible degradation products.11,12 The speed, the resolution and the sensitivity of UPLC separations, when combined with the high-speed scan rates of MS-QTOF (mass spectrometry quadrupole time-of-flight) detection, make the identification of degradation products more effective and the time required to characterize the DPs are shortened. Not surprisingly, this technique is gaining pre-eminence among the options for studying the intrinsic stability of drugs.13–19
Hence, the endeavor of the present work was to (i) carry out stress studies of the drug in hydrolysis, oxidation, photolysis and thermal (ii) selective and fast separation of the all DPs by using UPLC-PDA (iii) characterization of DPs by UPLC-QTOF-MS/MS experiments (iv) proposition of degradation pathway of the drug and its DPs and (v) prediction of in silico toxicity of all the DPs.
Experimental
Drugs and reagents
Pure MIR was purchased from Clearsyth Labs, Mumbai, India. LC-MS CHROMASOLV® grade methanol (MeOH) and acetonitrile (ACN) were procured from Sigma-Aldrich (Bangalore, India). Analytical reagent (AR) grade ammonium acetate, ammonium formate, formic acid, hydrochloric acid (HCl), sodium hydroxide (NaOH) and acetic acid were purchased from S.D. Fine Chemicals (Mumbai, India). AR grade H2O2 was purchased from Merck (Mumbai, India). HPLC grade water was obtained by using Milli-Q gradient system (Millipore, Bedford, MA, USA) and was used to prepare all solutions. ES Tuning Mix solution (Agilent Technologies, Palo Alto, CA, USA) was used as a MS/TOF calibrant.Instrumentations
All stability samples were analyzed by using an Acquity UPLC-H class system from M/s Waters having a flow-through needle design integral sample manager (SM-FTN), a bio quaternary gradient pump, an auto-injector and an in-line degasser. The column compartment with a temperature control and a photodiode array (PDA) detector was employed throughout the analysis. Chromatographic data was acquired using Empower 3 software.An Agilent 1200 series UPLC instrument (1290 Infinity, Agilent Technologies, USA) attached to a quadrupole time-of-flight (Q-TOF) mass spectrometer (6540 series, Agilent Technologies, USA) was used for the analysis of stressed degradation samples. The effluent of UPLC was directly attached with an electrospray ionization (ESI) source operated under positive mode with capillary voltage of 3500 V and fragmentor voltage of 170 V. Nitrogen was used as the drying (320 °C, 10 l min−1) and nebulizing (45 psi) gas. For collision-induced dissociation experiments, keeping MS1 static, the precursor ion of interest was selected using the quadrupole analyzer, and the product ions were analyzed using the TOF analyzer. Ultrahigh pure nitrogen gas was used as collision gas.
An ultra-sonicator from Power Sonic-405 (Hwashin Technology Co. Seoul, South Korea) and pH meter from pH tutor (Eutech Instruments, Singapore) were used to dissolve the sample and measure the pH of the mobile phase, respectively. The hydrolytic and thermal stress degradation studies were carried out using a high precision water bath and hot air oven equipped with digital temperature control of controlling the temperature within the range of ±2 °C and ±1 °C, respectively (Osworld scientific Pvt. Ltd. India). The stress photo degradation was carried out in a photostability chamber (Osworld OPSH-G-16-GMP series, Osworld scientific Pvt Ltd. India) capable of controlling the temperature and humidity within a range of ±2 °C and ±5% RH, respectively.
Stress degradation studies and its sample preparation
Stress studies were carried out on 500 μg ml−1 solution of MIR as per ICH recommended conditions of hydrolysis, oxidation, thermal and photolysis. Concentration of stressor was initiated with milder concentration followed stronger concentration so as to get sufficient degradation. As the drug was sparingly soluble in water and freely soluble in methanol, all the stress samples were prepared in a mixture of MeOH and the aqueous stressor (HCl, NaOH, water, H2O2) at ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50% v/v to the final concentration of 500 μg ml−1. Hydrolytic stress degradation study was carried out in 0.5 N NaOH, 1 N HCl and water at 80 °C for 2 h, 1 h and 48 h. For oxidative stress degradation, MIR was subjected to 15% H2O2 at room temperature for 8 h. Based on “A stress testing benchmarking study” found that pharmaceutical companies typically carry out the thermal degradation studies at ≥70 °C temperature. Hence in the present study, thermal degradation was carried out in solid state by exposing pure MIR in a Petri plate with a very thin layer to dry heat at 80 °C for 5 days.20 A photolytic stress study was carried out in solution and solid form at 40 °C in a photo stability chamber,9 equipped with an illumination bank made of light sources of a cool white fluorescent lamp designed for emitting significant radiation at 320 nm and a near UV fluorescent lamp with energy emission between 350 and 370 nm for providing an overall illumination of not less than 1.2 million lux hours and irradiation density of not less than 200 W m−2. A parallel set of the drug solutions was stored in dark at the same temperature to serve as control. Samples were prepared by filtering the solution through 0.22 μ filter paper prior to UPLC-PDA and UPLC-MS analysis. Samples were withdrawn at different time interval and diluted with mobile phase before injection. Both the acid and base degradation samples were neutralized with NaOH and HCl, respectively.
:50% v/v to the final concentration of 500 μg ml−1. Hydrolytic stress degradation study was carried out in 0.5 N NaOH, 1 N HCl and water at 80 °C for 2 h, 1 h and 48 h. For oxidative stress degradation, MIR was subjected to 15% H2O2 at room temperature for 8 h. Based on “A stress testing benchmarking study” found that pharmaceutical companies typically carry out the thermal degradation studies at ≥70 °C temperature. Hence in the present study, thermal degradation was carried out in solid state by exposing pure MIR in a Petri plate with a very thin layer to dry heat at 80 °C for 5 days.20 A photolytic stress study was carried out in solution and solid form at 40 °C in a photo stability chamber,9 equipped with an illumination bank made of light sources of a cool white fluorescent lamp designed for emitting significant radiation at 320 nm and a near UV fluorescent lamp with energy emission between 350 and 370 nm for providing an overall illumination of not less than 1.2 million lux hours and irradiation density of not less than 200 W m−2. A parallel set of the drug solutions was stored in dark at the same temperature to serve as control. Samples were prepared by filtering the solution through 0.22 μ filter paper prior to UPLC-PDA and UPLC-MS analysis. Samples were withdrawn at different time interval and diluted with mobile phase before injection. Both the acid and base degradation samples were neutralized with NaOH and HCl, respectively.
Results and discussion
Optimization of the chromatographic conditions
The method was developed using acetonitrile and the volatile buffer (ammonium acetate, 10 mM) so that the developed method could be transferred to mass spectrometry studies. A Waters CSH C18 column (100 × 2.1 mm, 1.7 μm) was found to be suitable for this analysis. The method was further optimized by varying the different selectivity factors such as pH of mobile phase, ratio of organic solvent, flow rate and column oven temperature. During scouting experiments, it was found that a few oxidative degradants were not resolved at pH 3 and 4. Finally, acceptable separation was achieved using ammonium acetate buffer with pH 5.0 and acetonitrile as organic modifier with a flow rate of 0.3 ml min−1 in a linear gradient elution mode. A linear flow gradient program was set as follows: (Tmin/% B): 0/10, 6/60, 8/60, 10/10. Column was equilibrated with 10 column volumes of mobile phase at the initial gradient composition prior to sample injection. All the stress samples were analysed using a PDA detector in a scan mode covering a range of 200–400 nm and final chromatogram was extracted at 248 nm to detect the peaks of all the degradation products. The same method was transferred for UPLC-MS studies by optimizing all the MS parameters as discussed in experimental section.Degradation behavior of MIR under various stress conditions
The stress degradation behavior of MIR was investigated by using UPLC-PDA method. The overlay of UPLC-PDA chromatogram is shown in Fig. 1. | ||
| Fig. 1 The overlay of UPLC-PDA chromatogram of (a) basic, (b) acidic, (c) oxidation and (d) neutral hydrolytic stress conditions. | ||
The drug showed degradation under hydrolytic and oxidative stress conditions, while it was found to be stable in photolytic and thermal stress conditions. There was no change observed in the color of solution as well as in solid form of the drug after exposing to photolytic stress conditions. The drug was degraded under base, acid and neutral hydrolytic stress conditions after refluxing in 0.5 N NaOH for 2 h, 1 N HCl for 1 h and water for 48 h. Overall, the drug forms four DPs (DP1 to DP4), two DPs (DP1 and DP5) and one DP (DP1) under basic, acidic and neutral hydrolytic stress conditions. The drug was extensively degraded after exposure to 15% H2O2 at room temperature for 8 h, forming in total, three DPs (DP1, DP6 and DP7). Proposed structures of all DPs and % degradation of the drug are listed in Scheme 1 and Table 1 respectively.
 | ||
| Scheme 1 Postulated structures of protonated degradation products of MIR. | ||
| Stress degradation conditions | Peak area of the drug | % Degradation of the drug | ||
|---|---|---|---|---|
| a Photolytic conditions was exposed to a total dose of 200 Wh m−2 of UV-illumination and 1.2 × 106 lux h of fluorescent light. | ||||
| Pure drug | 871309 |
0.0 | ||
| Hydrolysis | Acid | 1 N HCl, 80 °C 1 h | 704889 |
19.1 |
| Neutral | H2O, 80 °C, 48 h | 835585 |
4.1 | |
| Base | 0.5 N NaOH, 80 °C, 2 h | 677007 |
22.3 | |
| Oxidation | 15% H2O2 25 °C, 8 h | 762395 |
12.5 | |
| Photolytica | Neutral | H2O | 871387 |
0.0 |
| Solid | — | 871289 |
0.0 | |
| Thermal | Solid | — | 871389 |
0.0 |
UPLC-QTOF-MS/MS studies
 | ||
| Fig. 2 UPLC-ESI-MS/MS spectrum of [M + H]+ of (a) MIR (m/z 397) at 15 eV, (b) DP1 (m/z 257) at 10 eV, (c) DP2 (m/z 299) at 10 eV and (d) DP3 (m/z 373) at 15 eV. | ||
 | ||
| Scheme 2 Proposed fragmentation pathway of protonated MIR. | ||
| Degradation product | Retention time (min) | Molecular formula [M + H]+ | Calculated m/z | Observed m/z | Error (Δppm) |
|---|---|---|---|---|---|
| a DP: degradation products. | |||||
| MIR | 5.50 | C21H25N4O2S+ | 397.1639 | 397.1644 | −1.26 |
| DP1 | 4.36 | C16H21N2O+ | 257.1648 | 257.1639 | 3.50 |
| DP2 | 5.13 | C18H23N2O2+ | 299.1754 | 299.1745 | 3.01 |
| DP3 | 7.18 | C20H25N2O3S+ | 373.158 | 373.1583 | −0.80 |
| DP4 | 7.56 | C21H25N2O3S+ | 385.1580 | 385.1561 | 4.93 |
| DP5 | 4.03 | C16H21N2O+ | 257.1648 | 257.1652 | −1.56 |
| DP6 | 3.47 | C21H25N4O3S+ | 413.1642 | 413.1643 | −0.24 |
| DP7 | 3.88 | C21H27N4O4S+ | 431.1748 | 431.1758 | −2.32 |
| MIR and its DPs | Elemental composition | Calculated m/z | Observed m/z | Error (Δppm) |
|---|---|---|---|---|
| a DP: degradation product. | ||||
| MIR | C21H25N4O2S+ | 397.1639 | 397.1644 | −1.26 |
| C21H23N4OS+ | 379.1587 | 379.1572 | 3.96 | |
| C13H14N3OS+ | 260.0852 | 260.0839 | 4.99 | |
| C16H19N2+ | 239.1543 | 239.1539 | 1.67 | |
| C9H8NO+ | 146.0600 | 146.0599 | 0.68 | |
| C5H5N2OS+ | 141.0117 | 141.0113 | 2.84 | |
| C8H10N+ | 120.0808 | 120.0802 | 4.99 | |
| C4H5N2S+ | 113.0168 | 113.0163 | 4.42 | |
| DP1 | C16H21N2O+ | 257.1648 | 257.1639 | 3.50 |
| C16H19N2+ | 239.1543 | 239.1537 | 2.51 | |
| C8H10N+ | 120.0808 | 120.0803 | 4.16 | |
| C8H7+ | 103.0542 | 103.0540 | 1.94 | |
| C6H5+ | 77.0386 | 77.0383 | 3.89 | |
| DP2 | C18H23N2O2+ | 299.1754 | 299.1745 | 3.01 |
| C18H21N2O+ | 281.1648 | 281.1638 | 3.56 | |
| C10H12NO+ | 162.0913 | 162.0906 | 4.32 | |
| C8H10N+ | 120.0808 | 120.0804 | 3.33 | |
| DP3 | C20H25N2O3S+ | 373.1580 | 373.1583 | −0.80 |
| C20H23N2O2S+ | 355.1475 | 355.1489 | −3.94 | |
| C17H17N2O+ | 265.1335 | 265.1338 | −1.13 | |
| C16H21N2O+ | 257.1648 | 257.1638 | 3.89 | |
| C12H14NO2S+ | 236.074 | 236.0730 | 4.24 | |
| C9H8NO+ | 146.0600 | 146.0601 | −0.68 | |
| C8H10N+ | 120.0808 | 120.0812 | −3.33 | |
| DP4 | C21H25N2O3S+ | 385.1580 | 385.1561 | 4.93 |
| C21H23N2O2S+ | 367.1475 | 367.1461 | 3.81 | |
| C16H21N2O+ | 257.1648 | 257.1636 | 4.67 | |
| C13H14NO2S+ | 248.0740 | 248.0736 | 4.67 | |
| C9H8NO+ | 146.0600 | 146.0605 | −3.42 | |
| C8H10N+ | 120.0808 | 120.0807 | 0.83 | |
| C4H7OS+ | 103.0212 | 103.0217 | −4.85 | |
| DP5 | C16H21N2O+ | 257.1648 | 257.1652 | −1.56 |
| C16H19N2+ | 239.1543 | 239.1554 | −4.59 | |
| C8H10N+ | 120.0808 | 120.0811 | −2.50 | |
| C8H7+ | 103.0542 | 103.0547 | −4.85 | |
| C6H5+ | 77.0386 | 77.0389 | −3.89 | |
| DP6 | C21H25N4O3S+ | 413.1642 | 413.1643 | −0.24 |
| C21H25N4O2S+ | 397.1693 | 397.1688 | 1.26 | |
| C21H23N4O2S+ | 395.1536 | 395.1546 | −2.53 | |
| C21H23N4OS+ | 379.1587 | 379.1577 | 2.64 | |
| C14H17N4OS+ | 289.1118 | 289.1106 | 4.15 | |
| C16H21N2O+ | 257.1648 | 257.1655 | −2.72 | |
| C16H19N2+ | 239.1543 | 239.1550 | −2.93 | |
| C5H5N2O2S+ | 157.0066 | 157.0073 | −4.46 | |
| C8H10N+ | 120.0808 | 120.0802 | 4.99 | |
| C8H7+ | 103.0542 | 103.0546 | −3.88 | |
| DP7 | C21H27N4O4S+ | 431.1748 | 431.1758 | −2.32 |
| C21H25N4O3S+ | 413.1642 | 413.1644 | −0.48 | |
| C21H25N4O2S+ | 397.1693 | 397.1691 | 0.50 | |
| C21H23N4OS+ | 379.1587 | 379.1589 | −0.53 | |
| C17H17N2O+ | 265.1335 | 265.1331 | 1.51 | |
| C13H14N3OS+ | 260.0852 | 260.0850 | 0.77 | |
| C16H19N2+ | 239.1543 | 239.1547 | −1.67 | |
| C9H8NO+ | 146.06 | 146.0605 | −3.42 | |
| C8H10N+ | 120.0808 | 120.0805 | 2.50 | |
| C4H5N2S+ | 113.0168 | 113.0165 | 2.65 | |
The ESI-MS/MS spectrum of the [M + H]+ ion of DP1 (m/z 257, Rt = 4.36 min) is shown in Fig. 2(ii). A mass difference of 140 Da lower than the drug indicates that DP1 might be formed by the amide bond hydrolysis. It was further confirmed based on the MS/MS fragmentation pattern of [M + H]+ ion of DP1 and HRMS data. The spectrum shows the product ions at m/z 239 (loss of H2O from m/z 257), m/z 120 (loss of C8H11NO from m/z 257), m/z 103 (loss of NH3 from m/z 120) and m/z 77 (loss of C2H2 from m/z 120) (Scheme 3). The base peak at m/z 120, which was also observed in the MS/MS of protonated drug, indicates the presence of 2-phenylethenamine group in DP1. Further investigation of DP1 was done by comparing the MS/MS profile with the drug. It was remarkable to notice that the product ions at m/z 141 and 113 were absent in DP1, which indicates that 2-aminothiazole group was eliminated in DP1 during base hydrolysis. Based on all these data, DP1 was identified as 2-((4-aminophenethyl)amino)-1-phenylethanol. The most probable mechanism for the formation of DP1 could be acid or basic hydrolysis of amide bond, as shown in Scheme 6. The elemental compositions of product ions have been confirmed by accurate mass measurements (Table 3).
 | ||
| Scheme 3 Proposed fragmentation pathway for protonated DP1–DP5. | ||
Fig. 2(iii) shows the ESI/MS/MS spectrum of [M + H]+ ion (m/z 299, Rt = 5.13 min) of DP2. The accurate mass data shows the molecular formula of C18H23N2O2+, suggesting that sulfur and two nitrogens were eliminated from the drug. It displays three major product ions at m/z 281, 162 and 120. Loss of H2O to give the product ion at m/z 281 (Scheme 3), suggesting the presence of the hydroxyl group, similar to that of the drug. The base peak was observed at m/z 120 due to 2-phenylethenamine ion. The observed fragmentation of protonated DP2 is found to be highly compatible with the proposed structure, N-(4-(2-((2-hydroxy-2-phenylethyl)amino)ethyl)phenyl)acetamide and the elemental compositions of all the product ions have been supported by accurate mass measurements (Table 3). A probable mechanism for the formation of DP2 is shown in Scheme 6.
The alkaline degradant, DP3 was eluted at 7.18 min (Table 2) with its [M + H]+ peak at m/z 373. Its elemental composition, C20H25N2O3S+ shows that two nitrogen and one carbon were eliminated and one oxygen was added to the drug during alkaline hydrolysis. The ESI-MS/MS spectrum shows structure indicative fragment ions at m/z 355 (loss of H2O from m/z 373), m/z 265 (loss of C3H5SO From m/z 355), m/z 257 (loss of C4H4O2S from m/z 373), m/z 236 (loss of C8H9N from m/z 373), m/z 146 (loss of C8H9N from m/z 236) and m/z 120 (loss of C9H7NO from m/z 265) (Fig. 2(iv)). This DP3 fragmented to an ion of m/z 257, which is similar to the structure of DP1. This proves that DP3 was generated by the elimination of two nitrogen and a carbon of 2-aminothiazole group of the drug under base hydrolytic condition. The structure was characterized as 4-((4-(2-((2-hydroxy-2-phenylethyl)amino)ethyl)phenyl)amino)-4-oxobutanethioic S-acid, based on the product ions as shown in Scheme 3. A probable mechanism for the formation of DP3 may involve a series of steps as shown in Scheme 6. It includes a nucleophilic attack of hydroxyl anion on 2-aminothiazole moiety resulting in the formation of hydroxyl substituted 2-aminothiazole group followed by ring opening cascade and elimination of diamine moiety.
The degradation product DP4 at m/z 385 [M + H]+ was eluted at 7.56 min and its molecular formula (C21H25N2O3S+), suggests the addition of an oxygen and elimination of two nitrogen from the drug (Fig. 3(i) and Table 2). The ESI-MS/MS spectrum shows the product ions at m/z 367, 257, 248, 146, 120 and 103 (Table 3). All these data are highly compatible with the proposed structure, O-methyl 4-((4-(2-((2-hydroxy-2-phenylethyl)amino)ethyl)phenyl)amino)-4-oxobut-2-enethioate (Scheme 3). A probable mechanism for the formation of DP4 under hydrolytic condition is shown in Scheme 6.
 | ||
| Fig. 3 UPLC-ESI-MS/MS spectrum of [M + H]+ of (a) DP4 (m/z 385) at 15 eV, (b) DP5 (m/z 257) at 10 eV, (c) DP6 (m/z 413) at 10 eV and (d) DP7 (m/z 431) at 10 eV. | ||
The ESI-MS/MS spectrum of [M + H]+ ion (m/z 257) of DP5 was eluted before the drug at 4.03 min. It shows the product ions at m/z 239, 120, 103 and 77 (Fig. 3(ii)) and the spectrum is very much similar to that of DP1, suggesting that DP1 and DP5 are constitutional isomers that have same carbon skeleton and same functional groups but differ from each other in the location of the functional groups. Further, structure indicative product ion at m/z 120 (due to 2-phenylethenamine ion), confirms that hydroxyl group is at the second carbon from the phenyl group. All these data are highly compatible with the proposed structure, 1-((4-aminophenethyl)amino)-2-phenylethanol for DP5. A probable mechanism for the formation of DP5 is shown in Scheme 6. It may involve the formation oxonium by the protonation of an alcohol of DP1 under acidic conditions followed by elimination of H3O+ moiety. This results in the formation of an alkene as intermediate followed by the reaction of ‘pi’ bond with H+ (acidic medium) and H2O leads to the formation of DP5. The elemental compositions of DP5 and its product ions were confirmed by accurate mass measurements (Table 3).
The degradation product, DP6 (Rt = 3.47 min) was formed under oxidation conditions. The mass difference of 16 u between the protonated drug (m/z 397) and the protonated degradant (m/z 413) indicates an addition of oxygen from hydrogen peroxide to the drug. The ESI-MS/MS spectrum displays the product ions at m/z 397, 395, 379, 289, 257, 239, 157, 120 and 103 (Fig. 3(iii)). The product ions at m/z 379, 239 and 120 were similar to that of [M + H]+ of the drug. The characteristic ion at m/z 397 indicates that it was produced by the loss of oxygen from DP6, suggesting the possibility of N-oxide. It is reported that N-oxides are labile and undergo thermal decomposition and deoxygenation during collision induced dissociation.21 The structure of DP6 was further justified through its fragmentation pathway as shown in Scheme 4. Based on these data, the proposed structure was, 2-amino-4-(2-((4-(2-((2-hydroxy-2-phenylethyl)amino)ethyl)phenyl)amino)-2-oxoethyl)thiazole 3-oxide. A probable mechanism for the formation of DP6 can be explained by a nucleophilic addition of a hydroperoxide anion to tertiary nitrogen of thiazole ring followed by hydroxide elimination and abstraction of hydrogens by hydroxide anions resulting in the formation of N-oxide (Scheme 7). The elemental compositions of DP6 and its product ions were confirmed by accurate mass measurements (Table 3).
 | ||
| Scheme 4 Proposed fragmentation pathway for protonated DP6. | ||
DP7 was generated as a major degradation product under oxidative stress condition. Its [M + H]+ ion at m/z 431 with an elemental formula of C21H27N4O4S+, suggests that DP7 was formed by the inclusion of two oxygen atoms in the drug. As per the nitrogen rule, even molecular mass (430 Da) suggests even no. of nitrogen atoms in the structure, the nitrogen atoms were intact in the structure. The ESI/MS/MS spectrum shows the structure indicative product ions at m/z 413, 397, 379, 265, 260, 239, 146, 120 and 113 (Fig. 3(iv) and Scheme 5). The product ions at m/z 146 and 120 were also present in the MS/MS spectrum of drug, suggesting that possibility of an addition of hydroxyl group on the thiazole moiety. Based on all these data, DP7 was characterized as 2-(2-amino-4,5-dihydroxy-4,5-dihydrothiazol-4-yl)-N-(4-(2-((2-hydroxy-2-phenylethyl)amino)ethyl)phenyl)acetamide. A probable mechanism of the formation of DP7 is depicted in Scheme 7.
 | ||
| Scheme 5 Proposed fragmentation pathway for protonated DP7. | ||
 | ||
| Scheme 6 Probable mechanism of formation of DP1–DP5 under hydrolytic stress conditions. | ||
 | ||
| Scheme 7 Probable mechanism of formation of DP1, DP6 and DP7 under oxidative stress conditions. | ||
In silico toxicity of its degradation products
TOPKAT (Discovery Studio 2.5, Accelrys, Inc., San Diego, CA, USA) and DEREK (Nexus v2.0, Lhasa Ltd., Leeds, UK), established in silico toxicity prediction software tool, were employed to predict the potential toxicity of the drug and its degradation products.22 TOPKAT (Toxicity Prediction by Komputer Assisted Technology) employs rigorously developed and validated Quantitative Structure Toxicity Relationship (QSTR) models to predict toxicity in terms of probability values. The QSTR models in TOPKAT are developed by regression analysis for continuous endpoints and by discriminant analysis for categorical endpoints. TOPKAT exploits various two-dimensional molecular, electronic and spatial descriptors to develop the prediction models. In order to estimates the assurance in the prediction; TOPKAT applies the patented Optimal Predictive Space (OPS) during validation of the method. Probability values lower than 0.30 are considered as low probabilities for any toxicological end point while probability values greater than 0.70 are considered as high probabilities.23DEREK (Deductive Estimation of Risk from Existing Knowledge) is knowledge and rule based toxicity prediction tool. In order to predict toxicity it employs a set of rules that are developed from the collective knowledge obtained by analyzing literature and suggestions of the toxicologists worldwide. It includes more than 50 structural alerts for different toxicological endpoints in humans, other mammals and bacteria. Every structural alert is linked with a toxicophore (a substructure known or thought to be responsible for the toxicity). Final toxicity assessment for the query molecule is a result of a two-step process. In first step, the program verifies whether any alerts in knowledge base match toxicophore in the query molecule, and then in second step the reasoning engine determines the probability of the molecule's toxicity.24
The results of predicted toxicity profile for drug and all DPs using TOPKAT is given in Table 4. By evaluating the probabilities values it can be concluded that the toxicity profile of drug and its DPs are broadly similar with some exceptions. The drug and its degradation products show high probability values for FDA carcinogenicity male rat single vs. Mult (v3.1), while probabilities values for NTP carcinogenicity call (female rat) (v3.2) is pretty low. Degradation products such as DP1, DP3 and DP5 show high values for Ames Mutagenicity (v3.1) which indicates that these DPs have a potential to mutate DNA and form a tumors in humans or animals.
| Model | MIR | DP1 | DP2 | DP3 | DP4 | DP5 | DP6 | DP7 |
|---|---|---|---|---|---|---|---|---|
| NTP carcinogenicity call (male rat) (v3.2) | 0.465 | 0.965 | 0.365 | 0 | 0.459 | 0.314 | 0.904 | 0.195 |
| Ames mutagenicity (v3.1) | 0 | 0.987 | 0.493 | 0.838 | 0.129 | 0.986 | 0.002 | 0.095 |
| NTP carcinogenicity call (female rat) (v3.2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| NTP carcinogenicity call (male mouse) (v3.2) | 0 | 0.001 | 0 | 0 | 0.061 | 0.006 | 0 | 0 |
| NTP carcinogenicity call (female mouse) (v3.2) | 0.109 | 0.779 | 0.103 | 0.007 | 0.73 | 0.008 | 0.051 | 0 |
| FDA carcinogenicity male rat non vs. Carc (v3.1) | 1 | 0 | 0.962 | 1 | 0.91 | 0 | 1 | 0 |
| FDA carcinogenicity male rat single vs. Mult (v3.1) | 0.999 | 0.999 | 1 | 1 | 1 | 0.999 | 1 | 0 |
| FDA carcinogenicity female rat non vs. Carc (v3.1) | 0.999 | 0 | 0.994 | 0.993 | 0.032 | 0.015 | 0.977 | 0 |
| FDA carcinogenicity female rat single vs. Mult (v3.1) | 0.988 | 1 | 1 | 1 | 0.918 | 0.995 | 0.949 | 0.071 |
| FDA carcinogenicity male mouse non vs. Carc (v3.1) | 0.611 | 0.003 | 0.428 | 0.983 | 0.992 | 0 | 0.783 | 0 |
| FDA carcinogenicity male mouse single vs. Mult (v3.1) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| FDA carcinogenicity female mouse non vs. Carc (v3.1) | 1 | 1 | 1 | 0.007 | 1 | 0.53 | 0.999 | 0 |
| FDA carcinogenicity female mouse single vs. Mult (v3.1) | 0 | 0.051 | 0.156 | 0.998 | 0.013 | 0.878 | 0.286 | 0.999 |
| Weight of evidence carcinogenicity call (v5.1) | 0 | 0.002 | 0.897 | 0 | 0 | 0.005 | 0 | 0 |
| Developmental toxicity potential (DTP) (v3.1) | 0.858 | 1 | 0.034 | 0.292 | 0 | 0.987 | 0.505 | 0.958 |
| Rat oral LD50 (v3.1) (g kg−1) | 5.0 g kg−1 | 2.1 g kg−1 | 3.0 g kg−1 | 2.3 g kg−1 | 645.2 mg kg−1 | 695.6 mg kg−1 | 5.3 g kg−1 | 10 g kg−1 |
| Rat maximum tolerated dose – feed/water (v6.1) (mg kg−1) | 170.4 mg kg−1 | 21.9 mg kg−1 | 48.5 mg kg−1 | 7.2 mg kg−1 | 8.2 mg kg−1 | 12.7 mg kg−1 | 125.8 mg kg−1 | 10.0 mg kg−1 |
Table 5 shows the qualitative results for the drug and its DPs for skin sensitization end points obtained using DEREK software. According to DEREK assessment the drug is not skin sensitive, however, some of the DPs (DP1, DP3, DP4 and DP5) shows skin sensitization. The skin sensitization structural alert for DP1 and DP5 is aromatic primary and secondary amine while the structural alerts for DP3 and DP4 are thiol or thiol exchange agent and alpha, beta-unsaturated amide or precursor respectively (details given in Table 5).
| Drug and DPs | Skin sensitization | ||
|---|---|---|---|
| Structural alert |  |
 |
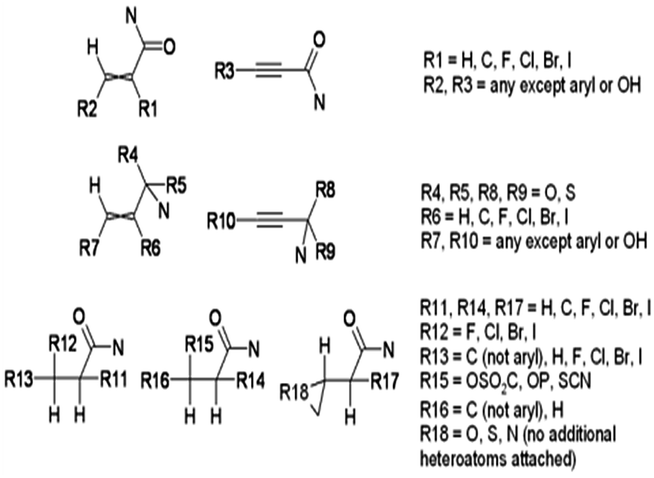 |
| Comment | This alert describes the skin sensitisation of aromatic amines and their N-protonated forms according to the toxicophores shown. In order to elicit a sensitisation response aromatic amines require transformation to a species capable of reacting with a skin protein nucleophilic group | Thiols (R2 = H) may react with skin protein either by nucleophilic attack at disulphide bridges or by a radical mechanism with thiol groups present in protein. Disulphides, trisulphides and sulphenamides (R2 or R3 = N) are electrophilic and thus may react with nucleophilic centres in skin proteins Alternatively, such compounds may be converted to the corresponding thiol by reaction with glutathione present in the skin | This alert describes the skin sensitisation of alpha, beta-unsaturated amides and precursors which interact with skin proteins via a Michael addition mechanism |
| MIR | NA | ||
| DP1 | √ | ||
| DP2 | NA | ||
| DP3 | √ | ||
| DP4 | √ | ||
| DP5 | √ | ||
| DP6 | NA | ||
| DP7 | NA | ||
Method validation
The developed stability indicating UPLC-PDA method of MIR was validated with respect to specificity, linearity, accuracy, precision and robustness as per ICH guideline Q2 (R1).25 Specificity was estimated on the stressed samples of MIR by using a PDA detector and the results showed that purity threshold was higher than purity angle which indicates that the drug peak is pure and proposed method is specific. Linearity was established at six concentrations in the range of 5–300 μg ml−1. The standard calibration curve was performed by least squares linear regression analysis. The linear regression equation and correlation coefficient (r2) were y = 9191x − 7806, and 0.999, respectively. The system suitability testing (SST) was performed by injecting five replicate of 100 μg ml−1 solution of the drug. The tailing factor (average: 1.04) and theoretical plate (average: 61210) were found well within the limits indicates that system is suitable to use. Accuracy was evaluated by spiking known concentration of the drug in triplicate at three level, low (10 μg ml−1), medium (100 μg ml−1) and high (200 μg ml−1) into the aliquot of stressed sample in triplicate. The recoveries of the added drug were obtained from the difference between peak areas of fortified and unfortified degraded samples. The percentage recovery range and RSD (relative standard deviation) values were found to be 98.0–101.0 and <1%, respectively (Table 6). Precision was determined in respect to repeatability and intermediate precision. Repeatability was evaluated with 3 determinations on the same day (n = 3) under the same operating conditions over a short interval of time. While intermediate precision was evaluated within-laboratories variations such as different days, different analysts and different UPLC columns (n = 3). The %RSD values were found to be below 1.0% for all the parameters indicating a method is precise (Table 7). Robustness of method was determined by intentionally changing in flow rate (±0.05 ml min−1), column temperature (±5 °C) and pH of mobile phase (±0.2). No significant change in assay value of MIR was observed by changing the chromatographic conditions which confirms that the developed method is robust.
| Amount added (μg ml−1) | Mean amount found (μg ml−1) ± SD | Recovery (%) |
|---|---|---|
| 10 | 10.12 ± 0.11 | 100.88 |
| 100 | 98.09 ± 0.13 | 98.27 |
| 200 | 201.69 ± 0.58 | 100.69 |
| Amount added (μg ml−1) | |||
|---|---|---|---|
| 10 | 100 | 200 | |
| Repeatability | |||
| Mean concentration | 10.09 | 98.22 | 201.74 |
| SD | 0.04 | 0.03 | 0.36 |
| %RSD | 0.43 | 0.03 | 0.18 |
| Intermediate precision | |||
| Day-1 (n = 3) | |||
| Mean concentration | 10.09 | 98.17 | 201.76 |
| SD | 0.07 | 0.26 | 0.81 |
| %RSD | 0.72 | 0.26 | 0.40 |
| Day-2 (n = 3) | |||
| Mean concentration | 10.08 | 98.16 | 201.51 |
| SD | 0.10 | 0.96 | 0.70 |
| %RSD | 0.94 | 0.98 | 0.35 |
| Day-3 (n = 3) | |||
| Mean concentration | 10.08 | 98.08 | 201.56 |
| SD | 0.08 | 0.93 | 0.65 |
| %RSD | 0.84 | 0.95 | 0.32 |
| Analyst-I | |||
| Mean concentration | 10.09 | 98.26 | 201.64 |
| SD | 0.03 | 0.37 | 0.67 |
| %RSD | 0.30 | 0.37 | 0.33 |
| Analyst-II | |||
| Mean concentration | 10.05 | 98.07 | 201.59 |
| SD | 0.06 | 0.12 | 0.50 |
| %RSD | 0.61 | 0.12 | 0.25 |
| Column-I | |||
| Mean concentration | 10.10 | 98.07 | 201.89 |
| SD | 0.10 | 0.12 | 0.77 |
| %RSD | 0.99 | 0.12 | 0.38 |
| Column-II | |||
| Mean concentration | 10.06 | 98.14 | 201.53 |
| SD | 0.08 | 0.09 | 0.48 |
| %RSD | 0.80 | 0.09 | 0.24 |
Conclusion
The degradation behavior of mirabegron under hydrolytic (acid, base and neutral), oxidative, photolytic and thermal stress conditions was studied as per ICH guidelines by developing the fast UPLC-PDA method. The drug underwent hydrolytic degradation under basic, acidic and neutral stressed conditions to yield four DPs (DP1, DP2, DP3 and DP4), two DPs (DP1 and DP5) and one DP (DP1), respectively. Three DPs (DP1, DP6 and DP7) were formed under oxidative stress conditions. All seven DPs were characterized unambiguously using on-line UPLC–ESI-MS/MS experiments combined with accurate mass measurements. Further, in silico toxicities were predicted for all DPs using TOPKAT and DEREK softwares. The results showed that degradants DP1, DP3 and DP5 revealed high probability for Ames mutagenicity which indicates that these DPs can cause mutations in the DNA and act as a carcinogen. DEREK software shows structure alert for DP1, DP3, DP4 and DP5 that are likely to cause skin sensitization. This study may also be useful in future investigation on identification and characterization of process-related impurities of the same class.Acknowledgements
The authors thank Dr Ahmed Kamal, Project Director, NIPER, Hyderabad and Dr M. Laksmi Kantham, Director, IICT for facilities and their cooperation. P.K is thankful to Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, New Delhi for the award of a Senior Research Fellowship.References
- T. M. Kessler, L. M. Bachmann, C. Minder, D. Lohrer, M. Umbehr, H. J. Schunemann and A. G. H. Kessels, PLoS One, 2011, 6, e16718 CAS.
- A. A. Bhide, G. A. Digesu, R. Fernando and V. Khullar, Int. Urogynecol. J., 2012, 23, 1345–1348 CrossRef PubMed.
- V. W. Nitti, S. Auerbach, N. Martin, A. Calhoun, M. Lee and S. Herschorn, J. Urol., 2013, 189, 1388–1395 CrossRef CAS PubMed.
- P. Tyagi, V. Tyagi and M. Chancellor, Expert Opin. Drug Saf., 2011, 10, 287–294 CrossRef CAS PubMed.
- M. Kashyap and P. Tyagi, Expert Opin. Drug Metab. Toxicol., 2013, 9, 617–627 CrossRef CAS PubMed.
- C. N. Bhimanadhuni and D. R. Garikapati, Am. J. PharmTech Res., 2012, 2, 564–571 Search PubMed.
- C. N. Bhimanadhuni and D. R. Garikapati, Der Pharma Chemica, 2013, 5, 55–60 CAS.
- R. V. Teijlingen, J. Meijer, S. Takusagawa, M. V. Gelderen, C. V. D. Beld and T. Usui, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 887, 102–111 CrossRef PubMed.
- ICH guideline, Q1B Photostability Testing of New Drug Substances and Products, International Conference on Harmonisation, IFPMA, Geneva, Switzerland, 1996 Search PubMed.
- ICH guideline, Q1A (R2) Stability Testing of New Drug Substances and Products, International Conference on Harmonisation, IFPMA, Geneva, Switzerland, 2003 Search PubMed.
- L. G. Valerio, Toxicol. Appl. Pharmacol., 2009, 241, 356–370 CrossRef CAS PubMed.
- L. G. Valerio, Hum. Genome, 2011, 5, 200 CrossRef CAS PubMed.
- P. D. Kalariya, M. V. N. K. Talluri, V. D. Gaitonde, P. S. Devrukhakar and R. Srinivas, J. Sep. Sci., 2014, 37, 2160–2171 CrossRef CAS PubMed.
- P. D. Kalariya, M. V. N. K. Talluri, P. N. Patel and R. Srinivas, J. Pharm. Biomed. Anal., 2015, 102, 353–365 CrossRef CAS PubMed.
- M. V. N. K. Talluri, N. R. Kandimalla, R. Bandu, D. Chundi, R. Marupaka and R. Srinivas, J. Pharm. Anal., 2014, 4, 107–116 CrossRef PubMed.
- T. Kosjek, S. Perko, E. Heath, B. Kralj and D. Zigon, J. Mass Spectrom., 2011, 46, 391–401 CrossRef CAS PubMed.
- M. V. N. K. Talluri, S. Dharavath, P. D. Kalariya, B. Prasanth and R. Srinivas, J. Pharm. Biomed. Anal., 2015, 105, 1–9 CrossRef PubMed.
- N. R. Ramisetti and R. Kuntamukkala, RSC Adv., 2014, 4, 23155–23167 RSC.
- N. R. Ramisetti and R. Kuntamukkala, New J. Chem., 2014, 38, 3050–3061 RSC.
- K. M. Alsante, L. Martin and S. W. Baertschi, Pharm. Technol., 2003, 27, 60–73 RSC.
- M. Holcapek, R. Jirasko and M. Lisa, J. Chromatogr. A, 2010, 1217, 3908–3921 CrossRef CAS PubMed.
- J. C. Dearden, J. Comput.-Aided Mol. Des., 2003, 17, 119–127 CrossRef CAS.
- K. Enslein, V. K. Gombar and B. W. Blake, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1994, 305, 47–61 CrossRef CAS.
- J. E. Ridings, M. D. Barratt, R. Cary, C. G. Earnshaw, C. E. Eggington, M. K. Ellis, P. N. Judson, J. J. Langowski, C. A. Marchant and M. P. Payne, Toxicology, 1996, 106, 267–279 CrossRef CAS.
- ICH guideline, Q2 (R1): Validation of Analytical Procedures: Text and Methodology, International Conference on Harmonization, IFPMA, Geneva, Switzerland, 2005 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |