DOI:
10.1039/C5RA01683E
(Paper)
RSC Adv., 2015,
5, 34956-34966
The quantitative detection of the uptake and intracellular fate of albumin nanoparticles†
Received
25th February 2015
, Accepted 9th April 2015
First published on
9th April 2015
Abstract
Little has been investigated about the intracellular fate of organic nanoparticles (NPs), which is important for the safety and drug delivery efficiency of NPs. In this study, to understand the NP cellular uptake and degradation characteristics, a quantitative method based on fluorescence resonance energy transfer (FRET) was developed and validated to detect the uptake and intracellular degradation of albumin NPs in MCF-7 cells. The effects of the crosslinking density and particle size on the intracellular uptake and degradation kinetics of albumin NPs were then systematically detected. The results indicated that the albumin NPs with higher crosslinking degrees could be internalized more quickly. With increasing NP diameter, the uptake number of NPs decreased, but the uptake NP weight increased due to the compensation of the increased weight of a single particle. The intracellular degradation results showed the NPs with a low crosslinking degree or a small diameter dissociated more quickly in cells, and the half-lives for the albumin NP dissociation were in the range of 35–79 h. These findings will provide fundamental but direct information for the optimal design and biomedical applications of NPs based on their intracellular fates, and the FRET method developed in this study can provide a novel and robust tool to track and monitor the NP intracellular fate.
Introduction
Due to the rapid development of materials science and nanotechnology, biodegradable nano-sized drug delivery systems (NDDSs, such as nanoparticles, micelles, liposomes, etc.) have been extensively investigated for disease diagnosis1–3 and treatment.4–6 Till now, large amounts of functional NDDSs7–13 have been proposed in labs or become pharmaceutical products, e.g. Abraxane®, Doxil®, Depocyt®, etc. but even the generally regarded safe carriers, such as poly-ε-caprolactone (PCL), poly(DL-lactic acid), poly(lactide-cocaprolactone), and poly(lactide-co-glycide) (PLGA) nanoparticles (NPs), showed toxicity at high concentrations in vitro.14,15 Therefore, it's very important to obtain detailed information and develop quantitative tools regarding the cellular uptake and the intracellular fate of biodegradable nanocarriers.6
Although the kinetics and mechanisms of cellular uptake of NDDSs have been well documented,6,16,17 to our best knowledge, very few reports focused on the intracellular fate of these nanocarriers.18–21 The lack of information on the intracellular fate, especially the intracellular degradation, not only made it difficult to design desired NDDSs, but also introduced excessive risk and sometimes even ethical problems for their future production and clinical application.22,23
Currently, the investigations of the NDDSs' intracellular fate were limited due to the immature quantitative detection method. For example, the method established for determination of PLGA NPs degradation in mice by detecting the molecular weight was limited by its low detection sensitivity and sample extraction difficulty,21 especially for the hydrophilic materials, which could hardly be extracted from the biological matrices efficiently. Another example is the coupled plasma mass spectrometry method developed for quantum dots intracellular elimination detection. This method may be applied only in the certain nano systems, which could release some elements or fragments with known molecular weights.24 Therefore, a quantitative method with high sensitivity and widely applicable range is critical and highly needed for determining the NDDSs' intracellular fate detection.
Fluorescence resonance energy transfer (FRET) involves the distance-dependent (<10 nm) transfer of energy from a fluorescent donor in its excited state to another excitable accepter through a long-range nonradioactive dipole–dipole interaction24 (Fig. 1A). As a powerful analytical technique for quantifying the distance between two fluorophores, it has been extensively and intensively utilized to interrogate changes in molecular conformation,25,26 interactions of biomolecular machinery,27,28 intracellular stability of polyplexes,29 etc.
 |
| | Fig. 1 The requirements of FRET phenomenon (A) and the degradation process of FRET-based nanoparticles (B). The FRET phenomenon requires a high quantum yield of donor, the overlap of donor emission spectrum and accepter absorption spectrum and the donor–accepter distance <10 nm. The degradation process of particles could be divided into the disassociation stage and the material hydrolysis stage. When the particle was disassociated, the donor–accepter distance was larger than 10 nm leading to the disappearance of FRET phenomenon. | |
In this report, the FRET theory was utilized and the NPs with FRET effect were designed to study the intracellular degradation of the NPs (Fig. 1B). In principle, in an intact NP, when the two dyes (the fluorescent donor and/or accepter labeled material), whose excitation and emission spectra overlap, are in sufficiently close proximity (<10 nm), the excited-state energy of donor molecule is transferred to the neighboring accepter molecule, resulting in a quenching of donor and an enhancement of accepter. If the NP is disassociated, the donor–accepter distance is larger than 10 nm, resulting the disappearance of FRET phenomenon. Therefore, a sensitive FRET-based method was developed and validated for the quantitative detection of NPs' intracellular degradation and fluorescein isothiocyanate (FITC) and rhodamine B isothiocyanate (RBITC) were selected as the fluorescent donor and accepter, respectively.
Albumin, a biodegradable, nontoxic and non-immunogenic protein, is an attractive material for NDDSs preparation,30 was selected as the building material for NP. Plenty of albumin nanocarriers30–33 have developed to improve their pharmacokinetic and target behaviors, and the commercial albumin-bound paclitaxel (Abraxane®) had already shown good safety and efficiency in the clinical application.34,35 It has been reported that the both clathrin- and caveolae-mediated pathway could contribute to the intracellular endocytosis of albumin NPs,36 but the intracellular fate and the factors influencing the uptake and degradation of them were still unclear. In this report, as a model platform, a FITC or RBITC labeled bovine serum albumin (BSA) NP library was developed with different crosslinking degrees and diameters to investigate the uptake and intracellular degradation kinetics of NPs. Furthermore, the NPs' uptake pathway was also determined in this study.
Results and discussion
Albumin NPs preparation and characterization
To investigate the effects of physicochemical properties on the uptake and intracellular fate of BSA NPs, a BSA NP library was developed with a series of crosslinking degrees and diameters, but comparable zeta potentials (−20 to −30 mV). These physicochemical properties are summarized in Table 1. The NPs with a series of crosslinking degrees from 41.9% to 91.1% (C40S70, C70S70 and C90S70) were obtained by reacting with different amounts of glutaraldehyde. The NPs (C40S70, C40S160 and C40S260) formulated with different albumin concentrations had comparable crosslinking degrees of about 42% but different diameters (71.8 nm, 157.4 nm and 261.9 nm). The fluorescence emission spectra of these NPs were shown in Fig. 2, and the highly quenching of donor and enhancement of accepter indicated the NPs possessed evident FRET effects, which guaranteed a significant fluorescence change even if only a tiny NPs disassociation occurred, thus providing a high sensitivity for the NPs disassociation detection. The FRET indices of NPs with various diameters were about 23%; those of NPs with different crosslinking degrees were in the range of 23–32% with larger indices being possessed by NPs of bigger crosslinking degrees, and this was perhaps because the chemical crosslink decreased the albumin molecular distances.
Table 1 Physicochemical properties of FRET-based BSA NPs
| |
Crosslinking degree (%) |
Diameter (nm) |
Zeta potential (mV) |
FRET index (%) |
| C40S70 |
41.9 ± 1.5 |
71.8 ± 0.1 |
−20.86 ± 1.53 |
23.6 ± 0.9 |
| C70S70 |
66.2 ± 2.1 |
72.0 ± 0.1 |
−24.09 ± 1.66 |
27.0 ± 2.8 |
| C90S70 |
91.1 ± 0.8 |
71.7 ± 1.4 |
−27.12 ± 1.57 |
31.9 ± 0.4 |
| C40S160 |
42.3 ± 1.0 |
157.4 ± 2.3 |
−23.58 ± 1.33 |
23.1 ± 0.3 |
| C40S260 |
41.8 ± 1.1 |
261.9 ± 1.2 |
−20.13 ± 1.05 |
23.4 ± 1.3 |
 |
| | Fig. 2 The fluorescence emission spectra of FITC-BSA (A), RBITC9.5-BSA (B) and the albumin NPs with various crosslinking degrees (C) and diameters (D). A, C and D: excited at 488 nm. B: excited at 548 nm. The quenching of fluorescence donor and enhancement of accepter indicated the albumin NPs possessed evident FRET effects. | |
Validation of the NPs degradation detection method
To validate the method developed for the NPs degradation determination, the NPs with various crosslinking degrees and diameters were incubated in trypsin buffers, and the disassociation fractions and hydrolysis degrees37 of NPs detected by the developed FRET-based method were compared with the results detected by the photometrical method38 and the pH-stat method.39 Different trypsin concentrations (6.6, 33, 66 and 100 µg mL−1, pH 8.0) were applied in this study, and the disassociation rates of C40S70 increased with the trypsin concentration as shown in Fig. 3A1–A4. The first order disassociation rate constants were calculated as shown in Table 2. When the trypsin concentration was set at 66 and 100 µg mL−1, comparing with those calculated by the photometrical method, significantly decreased disassociation rate constants were obtained by the FRET method; this is because when using the photometrical method, the intact NPs concentrations were underestimated for the rapidly decreased particle size brought in by the high trypsin concentrations. When the trypsin concentrations were set at 6.6 and 33 µg mL−1, no significant difference of the disassociation rate constants were observed between the two methods. As shown in Fig. 3B1–B4, similar results for the degree of hydrolysis (DH) were obtained by the FRET-based method and the pH-stat method: during the degradation process the values of protein DH reached the plateau rapidly, and the hydrolysis behaviors were independent on the trypsin concentration. These results indicate the FRET-based method works well for the NPs degradation detection, and this could also be concluded from the results of the other NPs (C70S70, C90S70, C40S160 and C40S260), which were shown in Fig. S1.†
 |
| | Fig. 3 Validation of the FRET-based method by detecting the albumin NPs degradation in trypsin buffers. (A) Disassociation fractions determined by the FRET method and the photometrical method. (B) Values of DH determined by the FRET method and the pH-stat method. The trypsin concentrations were set at 6.6 (A1, B1), 33 (A2, B2), 66 (A3, B3) and 100 µg mL−1 (A4, B4) (n = 3). | |
Table 2 Values of the disassociation rate constants of C40S70 in trypsin buffers detected by the photometrical method and the FRET-based method (n = 3)
| Trypsin (µg mL−1) |
k
a (h−1) |
k
b (h−1) |
|
Calculated from data detected by the FRET-based method.
Calculated from data detected by the photometrical method.
p < 0.05.
p < 0.01.
|
| 6.6 |
0.018 ± 0.003 |
0.017 ± 0.007 |
| 33 |
0.042 ± 0.016 |
0.049 ± 0.014 |
| 66 |
0.117 ± 0.015d |
0.179 ± 0.015c |
| 100 |
0.217 ± 0.044c |
0.302 ± 0.033c |
Cellular uptake pathway
Besides macropinocytosis, which was the major pathway for microparticle internalization,40 there are three types of pinocytosis for NPs: clathrin-mediated pathway, caveolae-mediated pathway and clathrin- and caveolae-independent pathway.16,41 To detect the mechanisms involved in the uptake of albumin NPs in the MCF-7 cells, the uptake of albumin NPs in the presence of chemical inhibitors was measured as shown in Fig. 4. A significant, but not complete, inhibition of uptake was observed in the presence of hypertonic medium, but no significant inhibition could be observed for all the NPs when using nystatin as the caveolae-mediated pathway inhibitor. The results showed that the uptake of the albumin NPs applied in this study was mainly via the clathrin-mediated endocytosis, and the uptake pathway was not influenced by the crosslinking degree and diameter of the albumin NPs.
Cellular uptake kinetics of albumin NPs
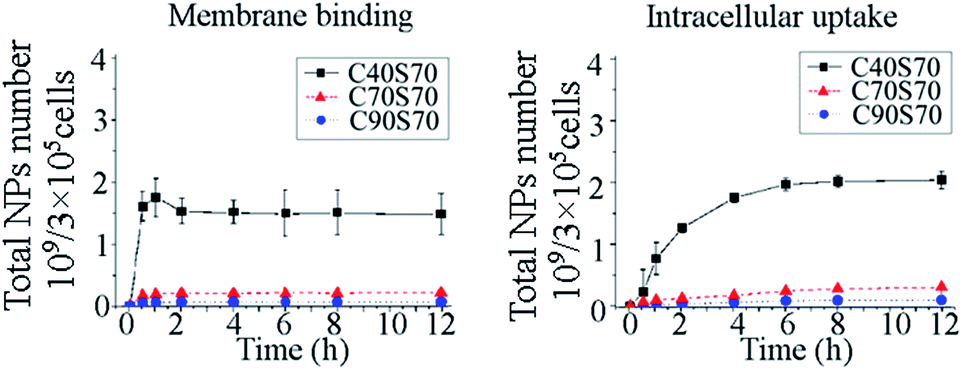
The membrane binding and intracellular uptake kinetics of the albumin NPs with different crosslinking degrees and diameters were detected in MCF-7 cells (Fig. 5).
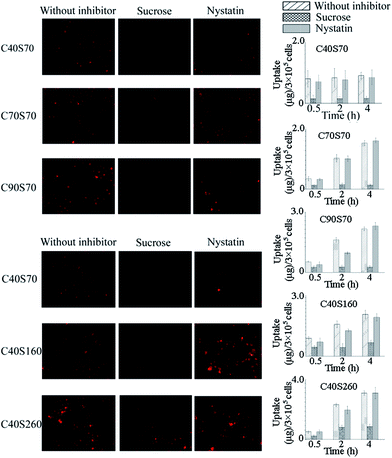 |
| | Fig. 4 Effects of inhibitors on the intracellular uptake of albumin NPs. The inhibitors applied were sucrose (450 mM) and nystatin (25 µg mL−1), which inhibited the clathrin-mediated and caveolae-mediated pathway, respectively. The inverted fluorescence microscope (Nikon Eclipse TI-5, Japan) was used for qualitative observation. The values of the intracellular uptake contents of NPs were obtained from the differences between the total cell-interacting and the membrane-binding data, which were detected at 37 °C and 4 °C, respectively (n = 3). | |
The effect of crosslinking degree on membrane binding and intracellular uptake of the albumin NPs are shown in Fig. 5. The results showed that the membrane binding was almost saturated within 1 h incubation, and NPs with higher crosslinking degree demonstrated a more efficient membrane binding. Compared with the intact NPs, the disassociated NPs in the membranes were negligible perhaps due to the lack of enzymes in the cell membranes. The intracellular uptake kinetics showed that the amount of intact albumin NPs increased within 6 h, and a slight decrease was observed from 6 to 12 h. Within the total incubation time of 12 h, the amount of disassociated NPs inside the cells increased gradually. The total amounts of albumin NPs in the MCF-7 cells increased within 6 h, at which time the uptake of C40S70, C70S70 and C90S70 plateaued, indicating saturation of uptake. The intracellular uptake kinetics showed the albumin NPs with higher crosslinking degrees (C90S70 and C70S70) were internalized more efficiently than those with a lower crosslinking degree (C40S70). A rapid hydrolysis of proteins disassociated from the NPs could be observed inside the cells as shown in Fig. 5.
 |
| | Fig. 5 The membrane binding and cellular uptake of the albumin NPs with different crosslinking degrees (C40S70, C70S70 and C90S70). The MCF-7 cells were incubated with albumin NPs (37.5 µg mL−1) at 37 °C and 4 °C to detect the total cell-interacting and membrane-binding amounts of NPs. The intracellular uptake amounts were obtained from the differences between the total cell-interacting and the membrane-binding data (n = 3). | |
The influences of diameter on the albumin NP intracellular uptake kinetics were shown in Fig. 6. For NPs with comparable crosslinking degrees but various diameters, a rapid saturation of the cell membrane-binding was observed, and with increasing diameter, a significant increase of binding could be obtained. As shown in the figure, a gradual increase of the intact NPs and the total amount of the albumin NPs in MCF-7 cells could be observed during the first 6 h, and the internalization increased with the increasing of particle size. The disassociated NPs inside the cells increased gradually within the period of 12 h. When the number of particles instead of their weight was used as the unit of measurement, it was interesting to find that compared with C40S160 and C40S260 the NPs with a smaller diameter (71.8 nm, C40S70) had the most efficient uptake (Fig. 7), concluding that the increase in diameter reduced the intracellular uptake particle number, but the weight of the intracellular particles was compensated by the increased weight of a single particle.
 |
| | Fig. 6 The membrane binding and cellular uptake of the albumin NPs with different diameters (C40S70, C40S160 and C40S260). The MCF-7 cells were incubated with albumin NPs (37.5 µg mL−1) at 37 °C and 4 °C to detect the total cell-interacting and membrane-binding amounts of particles. The intracellular uptake amounts were obtained from the differences between the total cell-interacting and the membrane-binding data (n = 3). | |
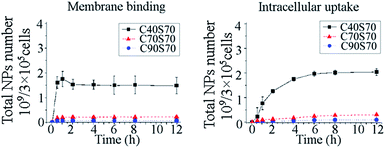 |
| | Fig. 7 The membrane binding and cellular uptake of the albumin NPs with different diameters (C40S70, C40S160 and C40S260) using number as the unit of measurement. The number of NPs was calculated from the formula 6m/(πR3ρ), where m was the weight of NPs, R was the diameter, and ρ was the density of NPs, which was determined as described by Ulbrich.44 | |
The cellular uptake kinetics of albumin NPs indicated the internalization of albumin NPs was controlled by the crosslinking degree and diameter of the NPs. As the nanoparticle–cell interface was the primary factor governing the interaction between particle and cell42 and the NPs with various crosslinking degrees offered comparable diameter and zeta potential, the deviations in the uptake behaviors were thought to be due to the different surface chemistry caused by the various crosslinking degrees. As shown, the uptake of albumin NPs was mediated by the clathrin-mediated pathway, which involves the ligand–receptor interaction and receptor-mediated endocytosis. It was thought the crosslink reaction reduced the ligand density of the albumin NPs and consequently affected their cellular uptake. Yuan and Zhang43 have theoretically elucidated the effects of ligand density and diameter on the kinetics of receptor-mediated endocytosis of NPs from thermodynamic aspect. It was revealed that because of the entropic penalty involved in ligand–receptor binding there existed an optimal condition (in terms of ligand density and diameter) at which the endocytic time is minimized. The time scale of full wrapping of NP was defined as l2/D, where l was a characteristic length, and D was the diffusivity of the receptor. Here, the values of l2 as the function of ligand density and diameter of the albumin NPs were calculated as shown in Fig. 8. As shown, when the ligand densities were larger than the optimal one, the increasing of crosslinking density could decrease the ligand density and consequently decrease the wrapping time of the albumin NP. When the diameter of albumin NPs were all larger than the optimal one, the increasing diameter increased the wrapping time and consequently decreases the NP's cellular uptake.
 |
| | Fig. 8 The influences of ligand density (ξl) and diameter of the albumin NPs on the values of l2, which was the characteristic length of the wrapping time scale of the NP (t ∼ l2/D) and was calculated as described by Yuan.43 (A) The diameter of albumin NPs were set at 72 nm, and the average ligand density of the MCF-7 cell membrane was set at 0.4, 0.7 and 1.0. (B) The ligand density was set at 0.7, 0.85 and 1.0, and the average ligand density of the MCF-7 cell membrane was set at 0.7. | |
The kinetics of disassociated NPs inside the MCF-7 cells was governed not only by the disassociation rate but also by the uptake of the NPs. Therefore, the degradation behaviors of these NPs were determined in absence of uptake in the following studies to elucidate the effects of crosslinking degree and diameter of albumin NPs on their intracellular degradation behaviors.
Intracellular degradation of albumin NPs
The intracellular degradation of albumin NPs was observed and detected based on the FRET phenomenon in absence of cellular uptake. As shown in Fig. 9A, the signal of channel D could hardly be observed within the uptake of 2 h. After 2 h, the NP-containing medium was replaced by fresh NP-free medium to terminate the particle uptake, and after the subsequent incubation for 8 h. Then a significant signal increasing from channel D (green fluorescence) could be observed for all the NPs (Fig. 9B), indicating the disassociation of particles occurred within the subsequent 8 h incubation.
 |
| | Fig. 9 The uptake (A) and the degradation (B) of albumin NPs observed by the confocal laser scanning microscope (Leica TCS SP5, Leica, Germany). The uptake was observed after incubation of MCF-7 cells with albumin NPs for 2 h, and the degradation was observed after the subsequent incubation of cells with the NPs-free medium for 8 h. | |
The quantitative results of the NP's intracellular degradation were shown in Fig. 10. For different amounts of NPs that were absorbed at the starting time of the degradation detection, the disassociated NPs were normalized by the initial NP content, and their disassociation fractions were obtained in the degradation detection. As shown in Fig. 10A1 and B1, no significant change in the amount of total cell-associated NPs could be observed during the degradation detection, indicating the exocytosis of the albumin NPs was negligible within the time period of 8 h. As shown in Fig. 10A2, a decrease of the disassociation fraction were observed when the crosslinking degree increased, indicating that the albumin NPs with a lower crosslinking degree are disassociated more efficiently. With comparable crosslinking degrees, the NPs with a small diameter of 71.8 nm (C40S70) were disassociated more quickly than those with diameters of 157.4 nm and 261.9 nm (C40S160 and C40S260), indicating the intracellular disassociation rate of the particles was inversely related to the particle size (Fig. 10B2). Quick and similar albumin hydrolysis behaviors of the NPs with different crosslinking degrees and diameters were observed in Fig. 10A3 and B3.
 |
| | Fig. 10 The degradation kinetics of albumin NPs with the series of crosslinking degrees (A) and diameters (B) within the uptake of 2 h and the subsequent degradation of 8 h in NPs-free medium. During the degradation of 8 h, the total amounts of cell-interacting NPs (M + N) had no significant changes, indicating the exocytosis of the albumin NPs was negligible within 8 h. The gradually increased disassociation fraction, which was calculated from N/(M + N), indicated the albumin NPs were degraded gradually in MCF-7 cells within the period of 8 h (n = 3). | |
The quantitative detection of NP degradation showed a gradual disassociation and quick hydrolysis of the particles in MCF-7 cells. The NPs with lower crosslinking degrees were disassociated more quickly in MCF-7 cells, indicating the extent of chemical crosslinking among the proteins was an important factor governing the NP's disassociation efficiency. The quick disassociation of the particles with smaller diameters may have resulted from the larger specific surface area and the consequently increased opportunities for the particle–enzyme interaction. Quick hydrolysis behaviors of the protein disassociated from NPs were observed in this study, indicating the disassociation was the rate-limiting step for the intracellular degradation of albumin NPs. The disassociation constants (k) of albumin NPs were obtained by fitting NPs' disassociation process using a first-order kinetic. The values of k and the half-lives of disassociation are shown in Table 3. When taking 5 times of half-lives as the washout period, after which more than 95% of NPs could be disassociated, the washout periods of albumin NPs applied in this study were in the range of 1–2.5 weeks. Although the data obtained in cell experiments could hardly represent in vivo results, it at least indicated the physicochemical properties of albumin NPs, such as their crosslinking degree and diameter, had an important impact on the clearance rate of NPs from the body and consequently on the safety of NPs.
Table 3 Fitting of the intracellular disassociation processes of albumin NPs using a first-order kinetic
| |
k (h−1) |
R
2
|
t
0.5 (h) |
Washout timea (h) |
|
The washout time was set as 5 half-lives, after which more than 95% of the albumin NPs could be disassociated.
|
| C40S70 |
0.0192 |
0.9928 |
36.134 |
180.67 |
| C70S70 |
0.0179 |
0.9839 |
38.619 |
193.095 |
| C70S90 |
0.0160 |
0.9955 |
43.443 |
217.215 |
| C40S160 |
0.0104 |
0.9854 |
66.359 |
331.795 |
| C40S2601 |
0.0081 |
0.9940 |
85.939 |
429.695 |
Experimental
Materials
BSA (96%), fluorescein isothiocyanate (FITC, 90%), rhodamine B isothiocyanate (RBITC, labeling efficiency with bovine albumin >70%) and cysteine (97%) were purchased from Sigma-Aldrich. Trypsin was purchased from Hualan Chemical. Paraformaldehyde (99%) was purchased from Shanghai Lingfeng Chemical Reagent Co., Ltd. RPMI-1640 medium, fetal calf serum and antibiotics (penicillin 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mL, streptomycin 10000 µg mL−1) were purchased from Hyclone Laboratories Inc. All other reagents were analytical grade and were purchased from Nanjing Chemical Reagent Co., Ltd.
000 units per mL, streptomycin 10000 µg mL−1) were purchased from Hyclone Laboratories Inc. All other reagents were analytical grade and were purchased from Nanjing Chemical Reagent Co., Ltd.
Albumin NPs preparation
FITC labeled BSA (FITC-BSA) and RBITC labeled BSA (RBITC9.5-BSA) were synthesized by dissolving 100 mg BSA in 4 mL of carbonate buffer and mixing with FITC (0.9 mg in 0.5 mL water) or RBITC (0.8 mg in 0.8 mL DMSO). After incubation for 2 h and dialysis against deionized water for 24 h, the fluorophore labeled BSA would be obtained after freeze drying.
Then an albumin NP library with a series of crosslinking degrees (C40S70, C70S70 and C90S70) and particle sizes (C40S70, C40S160 and C40S260) were developed with FITC-BSA and RBITC9.5-BSA using a disulfide bond reducing method established in our previous work.45 Briefly, the mixture of RBITC9.5-BSA and FITC-BSA with a weight ratio of 6:4 was dissolved in PBS buffer (pH 8.0) at 85 °C, and then a certain amount of cysteine was added to reach a final concentration of 5 mg mL−1. After dialysis against deionized water to remove the remnant cysteine, glutaraldehyde was added for the further crosslinking for 1 h. The remnant glutaraldehyde was removed by dialysis and the albumin NPs were obtained by lyophilization. The formulation parameters of the albumin NP library are shown in Table 4.
Table 4 The formulation of albumin NPs prepared using the disulfide bond reducing method
| |
Protein concentrationa (mg mL−1) |
Glutaraldehyde: protein (mg mg−1) |
|
The protein is the mixture of RBITC9.5-BSA and FITC-BSA with a weight ratio of 6:4.
|
| C40S70 |
1 |
0.04 |
| C70S70 |
1 |
0.1 |
| C70S90 |
1 |
0.2 |
| C40S160 |
2 |
0.04 |
| C40S2601 |
4 |
0.04 |
NP characterization
Particle size and zeta potential measurement.
The particle size and zeta potential of the NPs were determined using laser light scattering and a Zetaplus zeta potential analyzer (Brookhaven Instrument, USA), respectively.
Crosslinking degree determination.
After adding the same volume of acetic acid–sodium acetate buffer (pH 3.8) to the crosslinking reaction system, the NPs suspension was centrifuged at 8000 rpm for 30 min at 4 °C, and the supernatant was collected for glutaraldehyde detection. The crosslinking degrees of BSA NPs were determined using eqn (1).| |  | (1) |
where mguladd, mFITC-BSA and mRBITC9.5-BSA were the amounts (g) of glutaraldehyde, FITC-BSA and RBITC9.5-BSA added during the NPs preparation, respectively, and mglusupernatant was the amount (g) of glutaraldehyde in the supernatant after centrifugation at 8000 rpm for 30 min at 4 °C. Mglu and MBSA are the molecular weight of glutaraldehyde and BSA, respectively. The terms of nFITC-BSA and nRBITC9.5-BSA denoted the numbers of lysine in the molecule of FITC-BSA and RBITC9.5-BSA, which provided the primary amines with the capability of reacting with glutaraldehyde.
FRET index determination
The efficiency of the donor–accepter energy transfer of the NPs was reflected using a FRET index, which was calculated by eqn (2).46| |  | (2) |
where Fb and Db are the fluorescence intensity of NPs in the channel F and D; Fd and Dd are the fluorescence intensity of FITC-BSA in the channel F and D. The detection of signals in different channels was described in details in section “Sample fluorescence detection and data processing”.
FRET-based method development
In the FRET-based method, the fluorescence intensity was detected in different channels (D, F and A), and the excitation and emission of these channels were listed in Table 5.
Table 5 The excitation and emission measurements of signals in channel D, F and A
| Channel |
Excitation |
Emission |
| D |
Excitation of d (488 nm) |
Emission of d (517 nm) |
| F |
Excitation of d (488 nm) |
Emission of a (576 nm) |
| A |
Excitation of a (548 nm) |
Emission of a (576 nm) |
The samples of FITC-BSA (donor), RBITC9.5-BSA (accepter) and NPs were termed d, a and b, respectively. The fluorescence intensity of samples was termed using the letters describing the sample and the channel. For example, Dd was the fluorescence of the donor in the channel D.
In this study, the degradation of NPs was divided into two steps as shown in Fig. 1B: the disassociation of NPs and the hydrolysis of the protein disassociated from NPs. In the degradation process, the fluorescence intensity of samples was contributed by the intact NPs (M), the disassociated NPs (N) and the degree of hydrolysis37 of protein disassociated. According to the contribution of M, N and DH, signals in channel D, F and A (FD, FF and FA) can be described using the following equations.
| | | FD(M, N, DH) = FDb(M) + FDd(N) × fDd | (3) |
| | | FF(M, N, DH) = FFb(M) + FFd(N) × fFd | (4) |
| | | FA(M, N, DH) = FAb(M) + FAa(N) × fAa | (5) |
where
FDb,
FFb and
FAb were the fluorescence contribution of the intact NPs in channel D, F and A, and their standard curves were described by
eqn (6)–(8) using
α and
β as the slopes and intercepts of the standard curves.
FDd,
FFd and
FAa were the fluorescence intensities of the disassociated NPs in channel D, F and A and their standard curves are described by
eqn (9)–(11), where
n was the percentage of FITC-BSA in the NPs.
| | | FDd(N) = αDd × n × CN + βDd | (9) |
| | | FFd(N) = αFd × n × CN + βFd | (10) |
| | | FAa(N) = αAa × (1 − n) × CN + βAa | (11) |
where
CM and
CN are the concentrations of the intact NPs and the disassociated NPs. The slopes and intercepts in
eqn (6)–(11) could be obtained by plotting the fluorescence intensities in the corresponding channel against the concentrations of the samples.
Because of the self-quenching of FITC-BSA47,48 and RBITC9.5-BSA as shown in Fig. S2,† an increase of fluorescence can be attributed to the protein hydrolysis, and therefore the fluorescence intensities of the disassociated NPs (FDd, FFd and FAa) in eqn (3)–(5) were corrected by the DH terms (fDd, fFd and fAa). The DH terms could be determined by plotting the fluorescence intensities against the DH of FITC-BSA and RBITC9.5-BSA. Samples with different values of DH were prepared by digesting FITC-BSA and RBITC9.5-BSA (2.5 mg mL−1) in trypsin buffers at various concentrations. The DH of samples were detected using a pH-stat method.39 The relationships between fluorescence increasing and DH of proteins were shown in Fig. S3,† and it was found the DH terms of fDd, and fFd could be described using the linear equations (eqn (12) and (13)). As shown in Fig. S3,† a burst fluorescence increase was induced by the initial hydrolysis of RBITC9.5-BSA (DH of 0.49%), and after the burst increase, the fluorescence intensity reached a plateau. To keep consistent with the formats of eqn (12) and (13), a linear equation (eqn (14)) was still used to express fAa, but the slope (αDHAa) was set at 0 in the calculation.
| | | fDd = αDHDd × DH + βDHDd | (12) |
| | | fFd = αDHFd × DH + βDHFd | (13) |
| | | fAa = αDHAa × DH + βDHAa | (14) |
After substituting FDb, FFb, FAb, FDd, FFd, FAa, fDd, fFd and fAa into eqn (3)–(5), the unknown variables of M, N and DH could be obtained by solving the equation group using a sequential scanning method. A software program written in Visual Basic 6.0 was applied for the calculation.
Sample fluorescence detection and data processing
For the fluorescence detection, the fluorescence intensity of each sample in channel D, F and A were detected according to the excitation and emission method (Table 5) using the fluorescence spectrometer (RF 5301PC, SHIMADZU, Japan). The slit width of excitation and emission was set at 10 nm in this study. After substituting the fluorescence intensities into eqn (3)–(5), the intact NPs content (M), the disassociated NPs content (N) and the hydrolysis degrees of proteins could be obtained by solving the equation group using the program provided in the ESI.†
Method validation
To validate the method developed in this study, the degradation of albumin NPs in trypsin buffer was measured, and the disassociation fractions and values of DH calculated using the FRET-based method were compared with the results obtained by the photometrical method and pH-stat method.
FRET-based method.
NPs were suspended in the trypsin solution to a concentration of 500 µg mL−1 at 37 °C under shaking, and the pH was adjusted to 8.0. After various time intervals (0.25, 0.5, 1, 2, 4, 6, 8 and 12 h), 300 µL of the sample was withdrawn and diluted with PBS buffer (pH 8.0) to a volume of 3.0 mL for the fluorescence analysis. The disassociation fractions and DH of the NPs were obtained by solving eqn (3)–(5).
Photometrical method38.
The disassociation fractions calculated from the FRET-based method were compared with the results detected by the photometrical method. After suspending NPs in the trypsin solution, at the same time points mentioned above, 3 mL of the sample was withdrawn for photometrical detection at 565 nm. The NPs concentration was calculated using the calibration curve, and the disassociation fraction (DF) of the sample was calculated by eqn (15). The calibration curve was obtained by plotting the turbidity of the NPs at 565 nm against the NP's concentration.| |  | (15) |
pH-stat method39.
The values of DH calculated from the FRET-based method were compared with those detected by the pH-stat method. At the same time points mentioned above, 10 mL of the sample was withdrawn, and the pH of the sample was adjusted to 8.0 using NaOH solution (1 M). The DH of the sample was calculated using eqn (16).| |  | (16) |
where CNaOH and VNaOH are the concentration and the volume of NaOH solution consumed for pH adjustment; CN is the disassociated NPs concentration calculated by the photometrical method; V is the volume of the sample detected; α is the reciprocal of pK for amino groups of albumin (1.5);49,50 h is the total number of peptide bonds in BSA (0.0088 mol g−1 protein).
Cell culture
Human breast cancer MCF-7 cells were cultured in RPMI 1640 medium containing 10% fetal calf serum and 1% antibiotics (penicillin 10000 units per mL, streptomycin 10000 µg mL−1) at 37 °C and 5% CO2.
Cellular uptake pathway detection
For the uptake inhibition study, MCF-7 cells seeded in 24 well plates were treated with the chemical inhibitors for 0.5 h at 37 °C. Then the NPs were added to reach a concentration of 37.5 µg mL−1. After various incubation periods, the effects of the inhibitors on the particle uptake were obtained by both inverted fluorescence microscope observations and quantitative detection. The inhibitors applied in this study included nystatin (25 µg mL−1)37 and sucrose (0.45 M),51 which inhibited the caveolae- and clathrin-mediated pathway of endocytosis, respectively.
Cellular uptake kinetic detection
For detection of the intracellular uptake kinetics, the series of albumin NPs diluted by serum-free medium to a concentration of 37.5 µg mL−1 were incubated with MCF-7 cells in 24 well plates at 37 °C and 4 °C. After different time intervals (0.5, 1, 2, 4 and 6 h), the NP-containing medium was removed, and the cells were washed with cold PBS buffer (pH 8.0). To avoid the interference of Rayleigh scattering light in the fluorescence detection, which was caused by the intact cells, the cells had to be splitted, and in consideration that the chemical lysis buffer might interfere the fluorescence detection, the three freeze–thaw method was used for cell splitting.52 After splitting through three freeze–thaw cycles (−80 °C and 37 °C), the cells in each culture well were diluted with PBS buffer (pH 8.0) to a volume of 3 mL and stored at −20 °C until the fluorescence detection in channel D, F and A. After substituting the fluorescence intensities of these channels (FD, FF and FA) into eqn (3)–(5), the concentrations of intact and disassociated NPs and the hydrolysis degrees could be obtained by solving the equation group using a sequential scanning method as mentioned in the section “FRET-based method development”.
As internalization uptake was an energy-dependent process, only membrane binding occurred at 4 °C, while both binding and uptake took place at 37 °C. Therefore, the amount of cell associated nanoparticles at 37 °C was the total amount interacting with cells, and that at 4 °C represented the amount bound to the cell membranes.52–54 The difference between the amounts at 37 °C and 4 °C represents the intracellular uptake amount.
Intracellular degradation of albumin NPs
To quantify the NPs intracellular degradation, cells seeded in 24-well plates were incubated with the NPs diluted in the serum-free medium (37.5 µg mL−1) for 2 h. After incubation, the medium was replaced by the fresh RPMI 1640 medium, and this time point was set as the starting time (0 h) for the degradation detection. At different time points (such as 2, 4, 6 and 8 h), the medium was removed, and the cells were washed with cold PBS buffer (pH 8.0). After splitting through three freeze–thaw cycles and dilution with PBS buffer to a volume of 3 mL, the samples were stored at −20 °C until fluorescence detection as mentioned above.
Confocal laser microscope observation
The fluorescence signals of the cells in the channel D and channel A could be recorded using the confocal laser scanning microscope (Leica TCS SP5, Leica, Germany) after fixing the cells with 4% paraformaldehyde. The signals in the channel D and A were excited at 488 nm and 543 nm, respectively.
Conclusions
In this study, an albumin NP platform with controllable crosslinking degrees and diameters were prepared using FITC-BSA and RBITC9.5-BSA. The cellular uptake and the intracellular fate these albumin NPs were detected quantitatively through a FRET-based method. It was found the albumin NP binding with the cell membrane is rapid and saturated within 1 h. The albumin NPs entered MCF-7 cells via the clathrin-mediated endocytosis, which was not affected by the crosslinking degree and diameter of the NPs. However, these NP properties did affect the intracellular uptake of the albumin NPs. The cellular uptake of the albumin NPs increased with the increasing of crosslinking degree (in the range of 42–92%). The NP number of uptake decreased with the increasing of diameter. However, the cellular uptake increased as diameter increased in terms of NP weight. The albumin NPs cellular degradation behaviour was investigated in the MCF-7 cells. The results indicated that the exocytosis of the NPs was negligible within the 8 h. The disassociation rate of the albumin NPs were dependent on the crosslinking degree and diameter of the NPs: with the decreasing of crosslinking degrees and diameters, the disassociation rates increased. The disassociation half-lives of the albumin NPs were found in the range of 35–79 h. In MCF-7 cells, the proteins disassociated from the NPs would be hydrolyzed quickly. All these findings provide fundamental information not only for the biological effects and safety of albumin NPs, but also for controlling their properties and intracellular fates which are important for their future drug delivery applications. Furthermore, the FRET-based method developed in this study could provide an applicable tool for the investigation of NP's intracellular behaviour.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (Program no. ZJ11335 and no. JKPZ2013007), the Natural Science Foundation of Jiangsu Province (Program no. BK2012355), the National Natural Science Foundation of China (no. 81402878), and by the College Graduate Research and Innovation Plan of Jiangsu Province (Program no. CXZZ13_0327).
Notes and references
- J. S. Park, M. K. Cho, E. J. Lee, K. Y. Ahn, K. E. Lee, J. H. Jung, Y. Cho, S. S. Han, Y. K. Kim and J. Lee, Nat. Nanotechnol., 2009, 4, 259–264 CrossRef CAS PubMed.
- G. Morral-Ruiz, P. Melgar-Lesmes, C. Solans and M. J. Garcia-Celma, J. Controlled Release, 2013, 171, 163–171 CrossRef CAS PubMed.
- J. Zhu, Y. Lu, Y. Li, J. Jiang, L. Cheng, Z. Liu, L. Guo, Y. Pan and H. Gu, Nanoscale, 2014, 6, 199–202 RSC.
- R. Xing, G. Liu, J. Zhu, Y. Hou and X. Chen, Pharm. Res., 2014, 31, 1377–1389 CrossRef CAS PubMed.
- Y. Ding, Y. Y. Zhou, H. Chen, D. D. Geng, D. Y. Wu, J. Hong, W. B. Shen, T. J. Hang and C. Zhang, Biomaterials, 2013, 34, 10217–20227 CrossRef CAS PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- N. H. Cho, T. C. Cheong, J. H. Min, J. H. Wu, S. J. Lee, D. Kim, J. S. Yang, S. Kim, Y. K. Kim and S. Y. Seong, Nat. Nanotechnol., 2011, 6, 675–682 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Nat. Protoc., 2006, 1, 246–252 CrossRef CAS PubMed.
- R. Shrestha, M. Elsabahy, S. Florez-Malaver, S. Samarajeewa and K. L. Wooley, Biomaterials, 2012, 33, 8557–8568 CrossRef CAS PubMed.
- S. Li, J. Zheng, D. Chen, Y. Wu, W. Zhang, F. Zheng, J. Cao, H. Ma and Y. Liu, Nanoscale, 2013, 5, 11718–11724 RSC.
- J. Wu and C.-C. Chu, J. Mater. Chem. B, 2013, 1, 353–360 RSC.
- W. Hai, M. Sujuan, C. Xueqin, Z. Junwei, L. Ming, T. Jianxin, R. Fangfang, D. Yukou, P. Yue and G. Hongwei, Nanotechnology, 2014, 25, 195702 CrossRef PubMed.
- Z. Yu, R. M. Schmaltz, T. C. Bozeman, R. Paul, M. J. Rishel, K. S. Tsosie and S. M. Hecht, J. Am. Chem. Soc., 2013, 135, 2883–2886 CrossRef CAS PubMed.
- J. Wu, D. Yamanouchi, B. Liu and C.-C. Chu, J. Mater. Chem., 2012, 22, 18983–18991 RSC.
- R. P. Singh and P. Ramarao, Toxicol. Sci., 2013, 136, 131–143 CrossRef CAS PubMed.
- H. Gao, W. Shi and L. B. Freund, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9469–9474 CrossRef CAS PubMed.
- L. W. Zhang, W. Baumer and N. A. Monteiro-Riviere, Nanomedicine, 2011, 6, 777–791 CrossRef CAS PubMed.
- H. Wartlick, B. Spankuch-Schmitt, K. Strebhardt, J. Kreuter and K. Langer, J. Controlled Release, 2004, 96, 483–495 CrossRef CAS PubMed.
- T. Chernenko, C. Matthaus, L. Milane, L. Quintero, M. Amiji and M. Diem, ACS Nano, 2009, 3, 3552–3559 CrossRef CAS PubMed.
- O. Lunov, T. Syrovets, C. Rocker, K. Tron, G. U. Nienhaus, V. Rasche, V. Mailander, K. Landfester and T. Simmet, Biomaterials, 2010, 31, 9015–9022 CrossRef CAS PubMed.
- A. K. Mohammad and J. J. Reineke, Mol. Pharm., 2013, 10, 2183–2189 CrossRef CAS PubMed.
- H. M. Kipen and D. L. Laskin, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2005, 289, 696–697 CrossRef PubMed.
- P. A. Schulte and F. Salamanca-Buentello, Environ. Health Perspect., 2007, 115, 5–12 CrossRef.
- T. Föster, Ann. Phys., 1948, 2, 55–75 CrossRef PubMed.
- A. C. Ferreon, J. C. Ferreon, P. E. Wright and A. A. Deniz, Nature, 2013, 498, 390–394 CrossRef CAS PubMed.
- L. Baird, D. Lleres, S. Swift and A. T. Dinkova-Kostova, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15259–15264 CrossRef CAS PubMed.
- H. Kim, H. S. Afsari and W. Cho, J. Lipid Res., 2013, 54, 3531–3538 CrossRef CAS PubMed.
- S. Y. Kim, M. J. Lee, Y. R. Na, S. Y. Kim and E. G. Yang, J. Cell. Biochem., 2014, 115, 271–280 CrossRef CAS PubMed.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Eur. J. Pharm. Biopharm., 2006, 63, 156–165 CrossRef CAS PubMed.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 157, 168–182 CrossRef CAS PubMed.
- J. Han, Q. Wang, Z. Zhang, T. Gong and X. Sun, Small, 2014, 10, 524–535 CrossRef CAS PubMed.
- X. Ming, K. Carver and L. Wu, Biomaterials, 2013, 34, 7939–7949 CrossRef CAS PubMed.
- J. Li, Y. Di, C. Jin, D. Fu, F. Yang, Y. Jiang, L. Yao, S. Hao, X. Wang, S. Subedi and Q. Ni, Nanoscale Res. Lett., 2013, 8, 176 CrossRef PubMed.
- M. Satouchi, I. Okamoto, H. Sakai, N. Yamamoto, Y. Ichinose, H. Ohmatsu, N. Nogami, K. Takeda, T. Mitsudomi, K. Kasahara and S. Negoro, Lung Cancer, 2013, 81, 97–101 CrossRef PubMed.
- P. Y. Xing, J. L. Li, Y. Wang, X. Z. Hao, B. Wang, L. Yang, Y. K. Shi and X. R. Zhang, Chin. J. Cancer Res., 2013, 25, 200–205 Search PubMed.
- Y. Mo, M. E. Barnett, D. Takemoto, H. Davidson and U. B. Kompella, Mol. Vision, 2007, 13, 746–757 CAS.
- J. K. Lam, W. Liang, Y. Lan, P. Chaudhuri, M. Y. Chow, K. Witt, L. Kudsiova and A. J. Mason, J. Controlled Release, 2012, 158, 293–303 CrossRef CAS PubMed.
- K. Langer, M. G. Anhorn, I. Steinhauser, S. Dreis, D. Celebi, N. Schrickel, S. Faust and V. Vogel, Int. J. Pharm., 2008, 347, 109–117 CrossRef CAS PubMed.
- D. L. Lemos, A. L. Lawrence and A. J. Siccardi III, Aquaculture, 2009, 295, 89–98 CrossRef CAS PubMed.
- D. S. Kohane, Biotechnol. Bioeng., 2007, 96, 203–209 CrossRef CAS PubMed.
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2009, 78, 857–902 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- H. Yuan and S. Zhang, Appl. Phys. Lett., 2010, 96, 033704 CrossRef PubMed.
- K. Ulbrich, M. Michaelis, F. Rothweiler, T. Knobloch, P. Sithisarn, J. Cinatl and J. Kreuter, Int. J. Pharm., 2011, 406, 128–134 CrossRef CAS PubMed.
- L. Jiang, Y. Xu, Q. Liu, Y. Tang, L. Ge, C. Zheng, J. Zhu and J. Liu, Int. J. Pharm., 2013, 443, 80–86 CrossRef CAS PubMed.
- D. L. Graham, P. N. Lowe and P. A. Chalk, Anal. Biochem., 2001, 296, 208–217 CrossRef CAS PubMed.
- G. Hungerford, J. Benesch, J. F. Mano and R. L. Reis, Photochem. Photobiol. Sci., 2007, 6, 152–158 CAS.
- E. W. Voss Jr, C. J. Workman and M. E. Mummert, BioTechniques, 1996, 20, 286–291 CAS.
- M. Karamac and A. Rybarczyk, Pol. J. Food Nutr. Sci., 2008, 58, 351–357 CAS.
- D. Shi, Z. He and W. Qi, Process Biochem., 2005, 40, 1943–1949 CrossRef CAS PubMed.
- C. N. Antonescu, M. Diaz, G. Femia, J. V. Planas and A. Klip, Traffic, 2008, 9, 1173–1190 CrossRef CAS PubMed.
- Y. Li, J. Wang, Y. Gao, J. Zhu, M. G. Wientjes and J. L. Au, AAPS J., 2011, 13, 585–597 CrossRef CAS PubMed.
- N. Düzgüne and N. Shlomo, Adv. Drug Delivery Rev., 1999, 40, 3–18 CrossRef.
- K. D. Lee, S. Nir and D. Papahadjopoulos, Biochemistry, 1993, 32, 889–899 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01683e |
| ‡ Both authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.