
General solvent-free ionic liquid catalyzed C–N/C–C coupled cyclization to diverse dihydropyrimidinones and new organic materials: Langmuir–Blodgett film study†
Swapan Majumdar*a,
Jhinuk Dea,
Ajitesh Palb,
Indra Ghosha,
Ranendu K. Natha,
Sandip Chowdhuryc,
Dipanwita Royd and
Dilip K. Maiti*d
aDepartment of Chemistry, Tripura University, Suryamaninagar 799 022, India. E-mail: smajumdar@tripurauniv.in; Fax: +91-381-2374802; Tel: +91-381-2379070
bState Forensic Science Laboratory, Narsingarh, Agartala 799 015, India
cIndian Institute of Chemical Biology, 4, Raja S. C. Mullick Road, Kolkata 700 032, India
dDepartment of Chemistry, University of Calcutta, University College of Science, 92, A. P. C. Road, Kolkata-700 009, India. E-mail: dkmchem@caluniv.ac.in; Fax: +91-33-2351-9755; Tel: +91-33-2350-9937
First published on 2nd March 2015
Abstract
An ionic liquid catalyzed dual C–N/C–C coupled cyclization of a three component assembly is demonstrated to access 3,4-dihydropyrimidin-2(1H)-one (DHPM) analogues under solvent-free green conditions. Innovative new organic materials are introduced with pure and mixed DHPM using LB high surface pressure, AFM and photophysical studies.
Organic compounds possessing aggregation properties have attracted the attention of research, fabrication, production and business communities as futuristic materials for next generation electronic devices. Organic molecules are quite innovative components for designing materials for high-tech devices of ultimate sensitivity because of their structural diversity leading to much improved semiconducting, conducting, non-conducting, photoluminescence, storage and display performances relative to silicon, copper and other traditional ingredients.1–5 They are also lighter, more flexible, non-magnetic, biodegradable and inexpensive with respect to their inorganic counterparts. Benzimidazoles, imidazoles, porphyrins, and other N-heterocycles are utilized as the main building blocks for organic electronics.2 Introducing an ubiquitous framework dihydropyrimidinone (DHPM)6-bearing several nanoscale gluing interactions2b (E, Fig. 1) into the active research area is desirable to achieve new organic materials for fabricating innovative devices. With the current global awareness of developing environment friendly green industrial technologies,7 it is urgent to divert our attention from multistep organic syntheses into the nonhazardous multi-component strategy8c towards green synthesis7–10 of functional molecules through development of solvent-free8a non-metallic processes such as using ionic liquid (IL)9,10 as a green solvent cum catalyst, which found scant application.10 Recent medicinal research found DHPMs as active pharmaceutical agents for epilepsy (A, Fig. 1), antihypertensive calcium ion channel blocker (B), prostatic hyperplasia (C), cancer (D), fungal, viral, bacterial, inflammatory and cardiovascular diseases.6,11,12 The syntheses of valuable DHPMs were achieved by Biginelli11 and Biginelli-type coupling of aldehyde, urea/thiourea and β-dicarbonyl compounds using mineral acids, Lewis acid catalysts, nanoparticles, IL, solvent-free and/or catalyst-free conditions.12
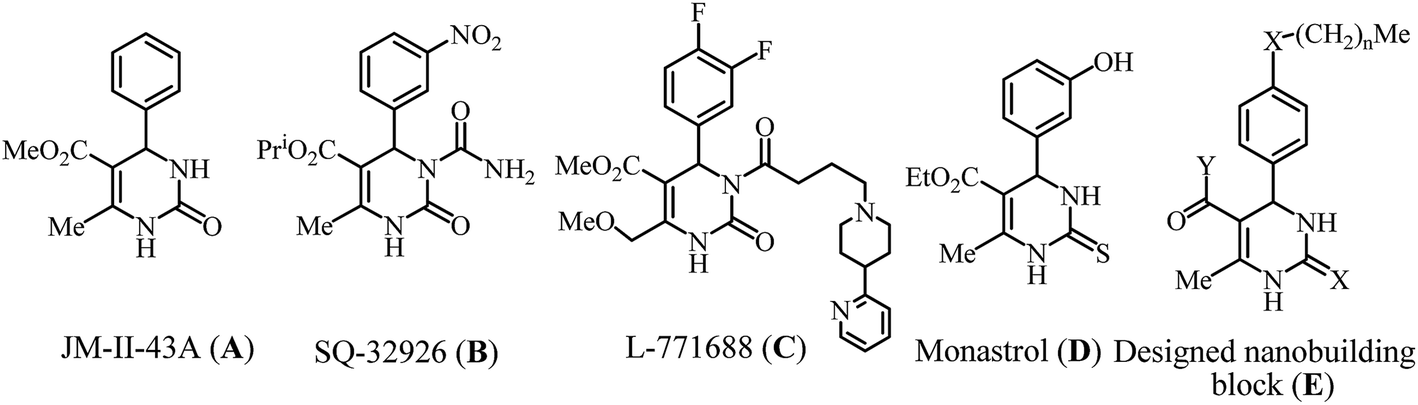 | ||
| Fig. 1 Medicinally significant and potential material DHPMs. | ||
Hitherto, all these methodologies come up with drawbacks using volatile organic solvents, excess quantity of IL, exhaustive usage of energy sources (e.g. high temperature), prolong reaction time, tedious catalyst preparation and isolation, formation of inevitable side products, and/or insufficient yield. With these difficulties in mind we developed a general solvent-free C–N/C–C coupled fundamental annulation process13 to DHPM analogues (Scheme 1) using protic ionic liquid as a catalyst. The preliminary results on LB study, aggregation property and surface morphology of the first ever DHPM-based materials are reported.
 | ||
| Scheme 1 A rapid C–N/C–C coupled annulation strategy to DHPMs. | ||
We decided to begin with an easily affordable protic IL catalyst such as imidazolium trifluoroacetate derivatives for developing a solvent-free green cyclization strategy of a three component assembly comprising benzaldehyde (R1 = Ph, 1a, Scheme 1), urea (X = O, 2a) and ethyl acetoacetate (R2 = OEt, 3a) to afford the desired DHPM (4a) and the successful annulation approach will be generalized and diversified through direct synthesis of varied functionalized and chiral DHPMs, their thio analogues and also the designed DHPMs of potential material properties (E, Fig. 1). Gratifyingly after a limited number of attempts we found rapid (30 min, entry 1, Table 1) conversion (100%) of the ingredients into the desired product 4a (entry 1) at moderate temperature (70 °C) using 10 mol% of PIL, 2-methyl-1-butyl imidazolium trifluoroacetate (I, Scheme 1). Interestingly progress of the reaction was ascertained simply by watching slow deposition of the solid product, which started within a couple of minutes. Comparable yield (89%) was obtained on reduction of catalyst loading from 10 to 5 mol% (entry 2). However the yield was reduced (entries 3 and 4) on further decrease of IL concentration (4–1 mol%), and no product was detected in absence of IL, heat or using Bu4NBr and BMImCl (entries 5–8). A plot for PIL (I) concentration vs. conversion of the starting material is included in the ESI.† However other ionic liquid such as BMImBF4 and BMImPF6 (ref. 9b) producedthe desired product with moderate yield (52–56%; entries 9 and 10) at high temperature. Thus, the results showed in Table 1 indicated that protic ionic liquid (I) offers best results than the other ionic liquid. We speculate that this is due to capability of protic ionic liquid I to activate carbonyl group11c,d of aldehydes which speeds up the iminium ion (X) formation with urea or thiourea and subsequent nucleophilic addition to the protic ionic liquid activated enolized-β-keto ester followed by cyclization (Z) and elimination of water results the expected DHPM (Scheme 2). Lacking of proton in other ionic liquids, carbonyls of aldehydes/β-dicarbonyls either not or less efficiently activated thus they required high temperature and long reaction time.
| Entry | Catalyst (mol%) | Temperature (°C) | Time (min) | Conversion (%) | Yieldb (%) |
|---|---|---|---|---|---|
| a 10 mmol scale.b Isolated yield.c 10 mol%.d No reaction at 70 °C. | |||||
| 1 | I (10) | 70 | 30 | 100 | 90 |
| 2 | I (5) | 70 | 30 | 100 | 89 |
| 3 | I (1) | 70 | 300 | 60 | 40 |
| 4 | I (4) | 70 | 300 | 90 | 81 |
| 5 | None | 70 | 300 | — | 0 |
| 6 | I (5) | 30 | 300 | — | 0 |
| 7 | Bu4NBrc | 100 | 30 | — | 0 |
| 8 | BMImClc | 100 | 30 | — | 0 |
| 9 | BMImBF4c | 100d | 60 | 90 | 56 |
| 10 | BMImPF6c | 100d | 60 | 80 | 52 |
 | ||
| Scheme 2 PIL (I) catalytic cycle for synthesis of DHPMs. | ||
The scope and versatility of the green strategy was successfully examined with a wide range of precursors towards rapid (10–90 min) access to functionalized DHPMs (4b–s, Fig. 2) with excellent yield (84–99%). A broad range of DHPMs possessing electron donating and electron withdrawing functionality substituted phenyl (4b–l), naphthyl (4m), conjugated π-bond (4n), and aliphatic substituents (4o–q) were synthesized. Acetyl acetone, flexible chiral aldehyde and thiourea were also smoothly underwent for the dual C–N/C–C coupling process to furnish corresponding DHPMs possessing acetyl group (4r and 4s), chiral amine-based 4t with about 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomer ratio and thione analogues (5a–e). To our delight the outstanding stereo-selection ability of the non-metallic PIL catalyst was observed to furnish optically pure single diastereomer 4u from structurally rigid chiral sugar aldehyde. Absolute stereochemistry (4u) was determined by NMR spectroscopic analysis (ESI†). In general, the reactions are very clean, generate no by product except water, furnish highly pure products after simple washing with water and require no chromatographic separation. However, poor solubility of urea hampered the precipitation process of the product leading to formation of impure material in few reactions. On dissolving urea in a little amount of water (0.5 mL) solved the problem without changing the reaction rate and yield. Alternatively crystallization in ethanol can be used to afford pure DHPM. The designed DHPM-bearing ester, long-chain alkoxy, donor HN–CO–NH, π-bond and aromatic moiety (4v) and its thio analogue (5f) were achieved under similar conditions.
:1 diastereomer ratio and thione analogues (5a–e). To our delight the outstanding stereo-selection ability of the non-metallic PIL catalyst was observed to furnish optically pure single diastereomer 4u from structurally rigid chiral sugar aldehyde. Absolute stereochemistry (4u) was determined by NMR spectroscopic analysis (ESI†). In general, the reactions are very clean, generate no by product except water, furnish highly pure products after simple washing with water and require no chromatographic separation. However, poor solubility of urea hampered the precipitation process of the product leading to formation of impure material in few reactions. On dissolving urea in a little amount of water (0.5 mL) solved the problem without changing the reaction rate and yield. Alternatively crystallization in ethanol can be used to afford pure DHPM. The designed DHPM-bearing ester, long-chain alkoxy, donor HN–CO–NH, π-bond and aromatic moiety (4v) and its thio analogue (5f) were achieved under similar conditions.
 | ||
| Fig. 2 Synthesized diverse DHPM analogues. | ||
We investigated the aggregation behaviour of 4v by using LB technique3 because it can efficiently be used for making films of controllable thickness, uniform surface and a high degree of orientational order of pure 4v and mixed DHPM–arachidic acid (AA). The mixed monolayers with AA3e was attempted for examining the valuable monolayer stability, second harmonic generation effects, pyroelectric, photochromic and intermolecular interactive properties. In our experiments, we observed that on spreading out of a dilute solution of pure 4v in chloroform (2 × 10−3 M) onto a pure water surface and compressing slowly resulted an initial rise of isotherm from zero to about 25 mN m−1 (Fig. 3A). A kink was observed and enhanced sharply to 55 mN m−1. The surface pressure versus area per molecule (π–A) isotherms of pure AA and 4v–AA mixed monolayers were executed (Fig. 3B) at the air–water interface. The abscissas of these figures represented the area of Langmuir film divided by the total number of the molecules, N = NEMT + NAA, where N4v and NAA are the numbers of 4v and AA molecules respectively. The pure AA isotherm was consistent with the behaviour expected from the previous measurements of this system. The isotherm of pure AA was resulted the limiting area of 0.20 nm2 indicating vertical standing of the long-chain fatty acids on water. The isotherm of pure 4v is very different to that of AA and no kink was observed. On mixing 4v with AA, the DHPM inserted into the monolayer, which was confirmed by changing in the lift off point and the existence of kink point in the mixed isotherm at about 25 mN m−1. The π–A isotherms of the 4v–AA mixed monolayer showed that the area/molecule enhanced with increasing mole fraction of 4v due to strong interaction between 4v–AA. The isotherms of the mixed films were resulted a shape that practically corresponds to the combination of those of individual components with small differences. The isotherms originated prominent kink points at higher mole fractions of 4v (0.7, 0.8, 0.9), which corresponded well with that of 4v. However, this kink point is practically absent at lower mole fractions of 4v (0.1, 0.3 and 0.5), which was similar to pure AA. Another noticeable feature of the isotherms of 4v–AA mixed mono-layers is that the isotherms did not converge at higher surface pressures suggesting retain of 4v in the mixed monolayer between the AA molecules. Thus the DHPM molecules didn't penetrate into the fatty acid monolayer, rather are located underneath the head groups of the densely packed AA monolayer at higher surface pressures. Determination of miscibility in the LB monolayer is very important to understand the basic interactions involved in the DHPM and AA molecules, which are responsible for the stability of the monolayer at the air–water interface. To analyze the degree of miscibility and thermodynamic nature of mixing of the binary components in the mixed LB, we examined the average area per molecule as a function of the film composition at different surface pressures (5, 10, 20, 30, 40 and 45 mN m−1, Fig. 3B). The nature of molecular interactions and also miscibility of the two components were examined by the quantitative analysis of the excess area (AE) of the mixed monolayer at the air–water interface.14a,b The excess area was obtained by comparing the experimentally observed average area per molecule (A12) of a mixed monolayer consisting of components 1 and 2 with that of an ideal mixed monolayer (Aideal):
| Aideal = A1N1 + A2N2 | (1) |
| AE = A12 − Aideal | (2) |
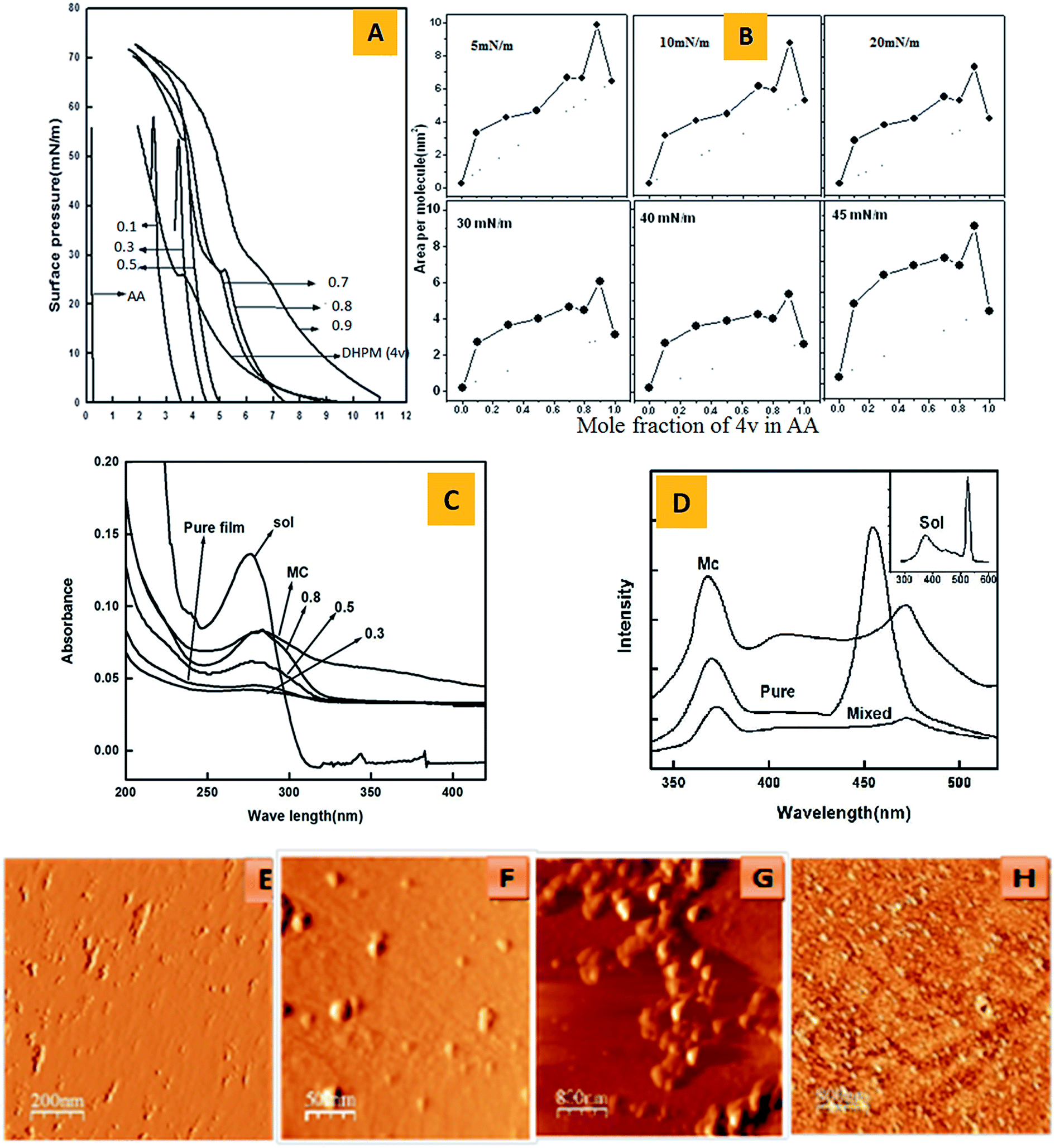 | ||
| Fig. 3 (A) Surface pressure (π) vs. area per molecule (A) isotherms of mixed monolayers of DHPM (4v) and AA at different mole fractions of 4v along with pure AA and 4v. (B) Plot of area/molecule versus mole fraction for mixed DHPM (4v)–AA monolayers on water sub-phase at various surface pressures. (C) UV-vis absorption spectra of DHPM CHCl3 solution (SOL), pure film of 4v microcrystal (MC) and 4v–AA mixed LB film at three different mole fractions namely, 0.3, 0.5, 0.8. (D) Emission spectra of DHPM (4v) in solution (SOL), 4v (pure), microcrystal (MC) and 4y/AA mixed LB film at a mole fraction of 0.5. (E) Atomic force microscope image of monolayer of AA, (F) pure film of DHPM 4v at 25 mN m−1, (G) pure film of DHPM 4v at 40 mN m−1, (H) mixed LB film of 4v–AA. | ||
The AFM imaging4 of LB monolayer of AA exhibited formation of an aggregated nanowire (panel E, Fig. 3). Similarly, the surface morphology of the pure DHPM (4v) monolayer transferred at 25 mN m−1 revealed formation of spherical aggregated material of about 100 nm dimension (panel F, Fig. 3). On enhancement of surface pressure to 40 mN m−1 changed the packing density of the 4v monolayer film leading to fabrication of grainy microstructure having a higher density domains (panel G, Fig. 3), which is an outstanding new observation. The AFM image of 4v–AA mixed LB film (panel H, Fig. 3) established the capability of the DHPM to aggregate with other component leading to construction of organic nanostructures materials, which has tremendous application during fabrication of organic electronic devices.
In conclusion, we have demonstrated a solvent-free, general, IL catalyzed dual C–N/C–C coupled-annulation strategy featuring operational simplicity, atom and energy efficiency, environmental safety, high conversion, fast reaction rate, excellent yield, and tolerance of functionalities and chirality to afford diverse DHPMs, their chiral and marcapto analogues. In addition, the UV-vis, fluorescence and AFM studies of pure and mixed thin films of the ubiquitous DHPM to innovative futuristic materials for organic electronics, and LB high surface pressure controlled development of outstanding aggregation property will find valuable application in chemical, material and medical sciences.
Acknowledgements
The authors gratefully acknowledge the financial grants of DST (no. SR/S1/OC-49/2010 and SR/NM/NS-29/2010) and CSIR (SRF), Govt. of India and also instrumental support from CRNN, CU. We are thankful to Mr Shantanu Chakraborty of our Dept. of Physics, TU for capturing AFM images.Notes and references
- (a) L.-L. Chua, J. Zaumseil, J.-F. Chang, E. C.-W. Ou, P. K.-H. Ho, H. Sirringhaus and R. H. Friend, Nature, 2005, 434, 194 CrossRef CAS PubMed; (b) S. R. Forrest and M. E. Thompson, Chem. Rev., 2007, 107, 923 CrossRef CAS; (c) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed; (d) J. E. Anthony, Nat. Mater., 2014, 13, 773 CrossRef CAS PubMed.
- (a) P. Pandit, N. Chatterjee, S. Halder, S. K. Hota, A. Patra and D. K. Maiti, J. Org. Chem., 2009, 74, 2581 CrossRef CAS PubMed; (b) D. K. Maiti, S. Halder, P. Pandit, N. Chatterjee, D. D. Joarder, N. Pramanik, Y. Saima, A. Patra and P. K. Maiti, J. Org. Chem., 2009, 74, 8086 CrossRef CAS PubMed; (c) C. J. Medforth, Z. Wang, K. E. Martin, Y. Song, J. L. Jacobsen and J. A. Shelnutt, Chem. Commun., 2009, 7261 RSC.
- (a) M. Puggelli, G. Gabrielli and G. Caminati, Thin Solid Films, 1994, 244, 1050 CrossRef CAS; (b) J. M. Chovelon, K. Wan and N. Jaffrezic-Renault, Langmuir, 2000, 16, 6223 CrossRef CAS; (c) J. Nath, R. K. Nath, A. Chakraborty and S. A. Husain, Surf. Rev. Lett., 2014, 21, 1450049 CrossRef; (d) B. Martín-García and M. M. Velázquez, Langmuir, 2014, 30, 509 CrossRef PubMed; (e) J. Collins, D. Funfschilling and M. Dennin, Thin Solid Films, 2006, 496, 601 CrossRef CAS PubMed.
- (a) J. J. Benítez, J. A. Heredia-Guerrero and A. Heredia, J. Phys. Chem. C, 2007, 111, 9465 CrossRef; (b) C. Storz, M. Badoux, C. M. Hauke, T. Solomek, A. Kühnle, T. Bally and A. F. M. Kilbinger, J. Am. Chem. Soc., 2014, 136, 12832 CrossRef CAS PubMed.
- (a) A. Ajayaghosh and S. J. George, J. Am. Chem. Soc., 2001, 123, 5148 CrossRef CAS; (b) T. E. Kaiser, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6719 CrossRef CAS PubMed.
- (a) A. D. Patil, N. V. Kumar, W. C. Kokke, M. F. Bean, A. J. Freyer, C. D. Brosse, S. Mai, A. Truneh, D. J. Faulkner, B. Carte, A. L. Breen, R. P. Hertzberg, R. K. Johnson, J. W. Westley and B. C. M. Potts, J. Org. Chem., 1995, 60, 1182 CrossRef CAS; (b) M. M. Ghorab, S. M. Abdel-Gawad and M. S. A. El-Gaby, Farmaco, 2000, 55, 249 CrossRef CAS; (c) B. ShivaramaHolla, B. SooryanarayanaRao, B. K. Sarojini and P. M. Akberali, Eur. J. Med. Chem., 2004, 39, 777 CrossRef PubMed; (d) N. Li, X.-H. Chen, J. Song, S.-W. Luo, W. Fan and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 15301 CrossRef CAS PubMed; (e) R. W. Lewis, J. Mabry, J. G. Polisar, K. P. Eagen, B. Ganem and G. P. Hess, Biochemistry, 2010, 49, 4841 CrossRef CAS PubMed; (f) R. Soni, G. Singh, R. Kaur, G. Kaur, R. K. Gill and J. Bariwal, Chem. Biol. Interface, 2014, 4, 163 Search PubMed.
- (a) Handbook of Green Chemistry and Technology, ed. J. Clark and D. Macquarrie, Blackwell Science, Oxford, UK, 2002 Search PubMed; (b) Sustainable Industrial Processes, ed. F. Cavani, G. Centi, S. Perathoner and F. Trifiró, Wiley-VCH, Weinheim 2009 Search PubMed; (c) M. Poliakoff and P. Licence, Nature, 2007, 450, 810 CrossRef CAS PubMed.
- (a) M. A. P. Martins, C. P. Frizzo, D. N. Moreira, L. Buriol and P. Machado, Chem. Rev., 2009, 109, 4140 CrossRef CAS PubMed; (b) K. Sanderson, Nature, 2011, 469, 18 CrossRef CAS PubMed; (c) K. S. Gayen, T. Sengupta, Y. Saima, A. Das, D. K. Maiti and A. Mitra, Green Chem., 2012, 14, 1589 RSC; (d) A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083 CrossRef PubMed.
- (a) M. Freemantle, An Introduction to the Ionic Liquids, RSC Publishing, London, 2009 Search PubMed; (b) J. Peng and Y. Deng, Tetrahedron Lett., 2001, 42, 5917 CrossRef CAS; (c) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS PubMed; (d) S. A. Dharaskar, K. L. Wasewar, M. N. Varma, D. Z. Shende and C. K. Yoo, Ind. Eng. Chem. Res., 2014, 53, 19845 CrossRef CAS.
- (a) B. C. Ranu and R. Jana, Eur. J. Org. Chem., 2006, 3767 CrossRef CAS; (b) A. K. Chakraborti, S. R. Roy, D. Kumar and P. Chopra, Green Chem., 2008, 10, 1111 RSC; (c) S. Majumdar, M. Chakraborty, D. K. Maiti, S. Chowdhury and J. Hossain, RSC Adv., 2014, 4, 16497 RSC; (d) S. Majumdar, J. De, A. Chakraborty and D. K. Maiti, RSC Adv., 2014, 4, 24544 RSC.
- (a) P. Biginelli, Chem. Ber., 1891, 24, 1317 CrossRef; (b) P. Biginelli, Chem. Ber., 1891, 24, 2962 CrossRef; (c) P. Biginelli, Chem. Ber., 1893, 26, 447 CrossRef; (d) P. Biginelli, Gazz. Chim. Ital., 1893, 23, 360 Search PubMed; (e) B. Seiller, C. Bruneau and P. H. Dixneuf, Synlett, 1995, 707 CrossRef CAS PubMed.
- (a) J. S. Yadav, B. V. S. Reddy, R. Srinivas, C. Venugopal and T. Ramalingam, Synthesis, 2001, 1341 CrossRef CAS PubMed; (b) B. C. Ranu, A. Hajra and S. S. Dey, Org. Process Res. Dev., 2002, 6, 817 CrossRef CAS; (c) A. Dondoni, A. Massi, S. Sabbatini and V. Bertolasi, J. Org. Chem., 2002, 67, 6979 CrossRef CAS PubMed; (d) C. D. Bailey, C. E. Houlden, G. L. J. Bar, G. C. Lloyd-Jones and K. I. Booker-Milburn, Chem. Commun., 2007, 2932 RSC; (e) J. M. Goss and S. E. Schaus, J. Org. Chem., 2008, 73, 7651 CrossRef CAS PubMed; (f) J.-P. Wan and Y.-J. Pan, Chem. Commun., 2009, 2768 RSC; (g) G. C. Nandi, S. Samai and M. S. Singh, J. Org. Chem., 2010, 75, 7785 CrossRef CAS PubMed; (h) S. R. Roy, P. S. Jadhavar, K. Seth, K. K. Sharma and A. K. Chakraborti, Synthesis, 2011, 2261 CAS; (i) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156 CrossRef CAS PubMed; (j) R. Wang and Z.-Q. Liu, J. Org. Chem., 2012, 77, 3952 CrossRef CAS PubMed; (k) L. Zhang, Z. Zhang, Q. Liu, T. Liu and G. Zhang, J. Org. Chem., 2014, 79, 2281 CrossRef CAS PubMed; (l) E. Kolvari, N. Koukabi and O. Armandpour, Tetrahedron, 2014, 70, 1383 CrossRef CAS PubMed; (m) H. G. O. Alvim, T. B. Lima, A. L. de Oliveira, H. C. B. de Oliveira, F. M. Silva, F. C. Gozzo, R. Y. Souza, W. A. da Silva and B. A. D. Neto, J. Org. Chem., 2014, 79, 3383 CrossRef CAS PubMed.
- (a) K.-T. Yip, M. Yang, K.-L. Law, N.-Y. Zhu and D. Yang, J. Am. Chem. Soc., 2006, 128, 3130 CrossRef CAS PubMed; (b) D. Dhara, K. S. Gayen, S. Khamarui, P. Pandit, S. Ghosh and D. K. Maiti, J. Org. Chem., 2012, 77, 10441 CrossRef CAS PubMed; (c) S. Ghosh, S. Khamarui, S. Gayen and D. K. Maiti, Sci. Rep., 2013, 3, 2987 Search PubMed.
- (a) A. W. Adamson and A. P. Gast, Physical Chemistry of Surfaces, John Wiley & Sons, New York, 6th edn, 1997 Search PubMed; (b) A. K. Dutta, T. N. Misra and A. J. Pal, Langmuir, 1996, 12, 459 CrossRef CAS; (c) H. C. Ko, S. Kim, H. Lee and B. Moon, Adv. Funct. Mater., 2005, 15, 2005 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01618e |
| This journal is © The Royal Society of Chemistry 2015 |