
Palladium-Schiff-base-silica framework as a robust and recyclable catalyst for Suzuki–Miyaura cross-coupling in aqueous media†
Tahshina Beguma,
Manoj Mondala,
Pradip K. Gogoi*a and
Utpal Bora*ab
aDepartment of Chemistry, Dibrugarh University, Dibrugarh 786004, Assam, India. E-mail: dr.pradip54@gmail.com; utbora@yahoo.co.in; ubora@tezu.ernet.in
bDepartment of Chemical Sciences, Tezpur University, Napaam, Tezpur 784028, Assam, India
First published on 10th April 2015
Abstract
A silica supported palladium catalyst, Pd@imine–SiO2 was prepared by immobilizing Pd(OAc)2 onto silica gel through coordination of imine, generated via Schiff-base condensation between 3-aminopropyltriethoxysilane (APTES) functionalized silica gel and salicylaldehyde. The prepared catalyst was characterized by FT-IR, BET surface area measurements, XRD, SEM-EDX, EDS-mapping and ICP-AES analysis. The imine-based catalyst exhibited excellent activity in the Suzuki–Miyaura cross-coupling reactions of aryl bromides with arylboronic acids in iPrOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature. The reaction proceeds under mild reaction conditions and the catalyst is recyclable, thus offering an environmentally benign alternative to the existing protocols.
:1) at room temperature. The reaction proceeds under mild reaction conditions and the catalyst is recyclable, thus offering an environmentally benign alternative to the existing protocols.
Introduction
Compounds containing carbon–carbon bonds have found extensive applications in the synthesis of natural products, agrochemicals, pharmaceuticals, and polymers.1 Among the various methods the palladium-catalyzed Suzuki–Miyaura reaction developed for the linkage of two similar/dissimilar aryl fragments via the cross-coupling of aryl halide and arylboronic acid has became the most dominant method for synthesizing homo-aryl and biaryl derivatives.2 Various palladium based catalysts ligated with simple tertiary phosphines, hemilabile-type phosphines, sterically crowded biphenyl-type phosphines,2d,3 imine,4 NHC,5 amine,6 bisamides,7 triazole,8 Schiff-base,9 acetanilide,10 and oxime-ether11 have been commonly used for Suzuki–Miyaura cross coupling reaction. However, the practical application of homogeneous catalyst in pharmaceutical industry remains challenging because the expensive catalysts are not easily reusable, hard to separate from reaction mixture (a frequent industrial concern) and often contaminates the product.12 Therefore, in order to meet regulatory standards, the methods for removal of these toxic metals from active pharmaceutical ingredients need to be highly efficient.13 Therefore, the use of heterogeneous catalyst seems promising and advantageous over homogenous catalysts in terms of minimizing contamination of the products, easy isolation, reusability and long-term stability of the solid catalyst.Nowadays, many effective solid supports are available that are easily accessible and often provides effective platform for heterogeneous catalysis. Silica, due to its low cost, easy accessibility, high thermal stability, excellent porosity14 and large surface area received utmost attention compared to other effective supports like alumina, poly(vinyl chloride), MCM-41,15 SBA-15,16 polymeric ionic liquid17 etc. Moreover, the highly active surface-silanol groups present in the silica promotes the effective anchoring of organic ligands onto its surface,18 and subsequently the immobilization of the metal via covalent bond on the support. Till date, noticeable advances have been carried out in synthesizing novel and efficient silica-supported heterogeneous palladium catalyst to perform Suzuki–Miyaura reaction.14,19 However, many of these methods still uses toxic organic or biphasic co-solvents, elevated reaction temperature and/or require high catalyst loading with large excess of boronic acids.14,18b,20 In addition, many of these heterogeneous catalysts suffer from the problem concerning poor recyclability due to certain unwanted inactivation processes, such as aggregation and growth of less reactive large palladium particles. Therefore, there is a need to develop novel recoverable catalytic system that can be efficiently reused under mild reaction condition while retaining the original activity of the homogeneous catalytic centers.
In coordination chemistry and catalysis, Schiff-bases are considered as an important class of N-based ligands, as they possess high selectivity and sensitivity towards metal ions and are well regarded as chelating multidentate ligands. However, in homogeneous catalysis the palladium Schiff-base complexes often get deactivated due to the formation of dimeric peroxo- and μ-oxo species.21 To overcome these drawbacks, researchers have devised various immobilized palladium based catalyst as efficient and recyclable catalyst for the Suzuki–Miyaura coupling of a variety of electronically diverse aryl halides under ambient reaction condition.18b,19b,20b,22
Through this communication, we wish to report the synthesis of a novel salicylaldehyde-based Schiff-base-palladium complex immobilized on silica support. The catalyst was found to be highly efficient in the Suzuki–Miyaura cross coupling reaction in aqueous solution at room temperature and could be recovered by simple filtration reused up to eight consecutive cycles without significant loss in activity.
Results and discussion
Synthesis and characterization of the materials
The palladium complex was synthesized in three steps following a previously reported procedure and is summarized in Scheme 1.23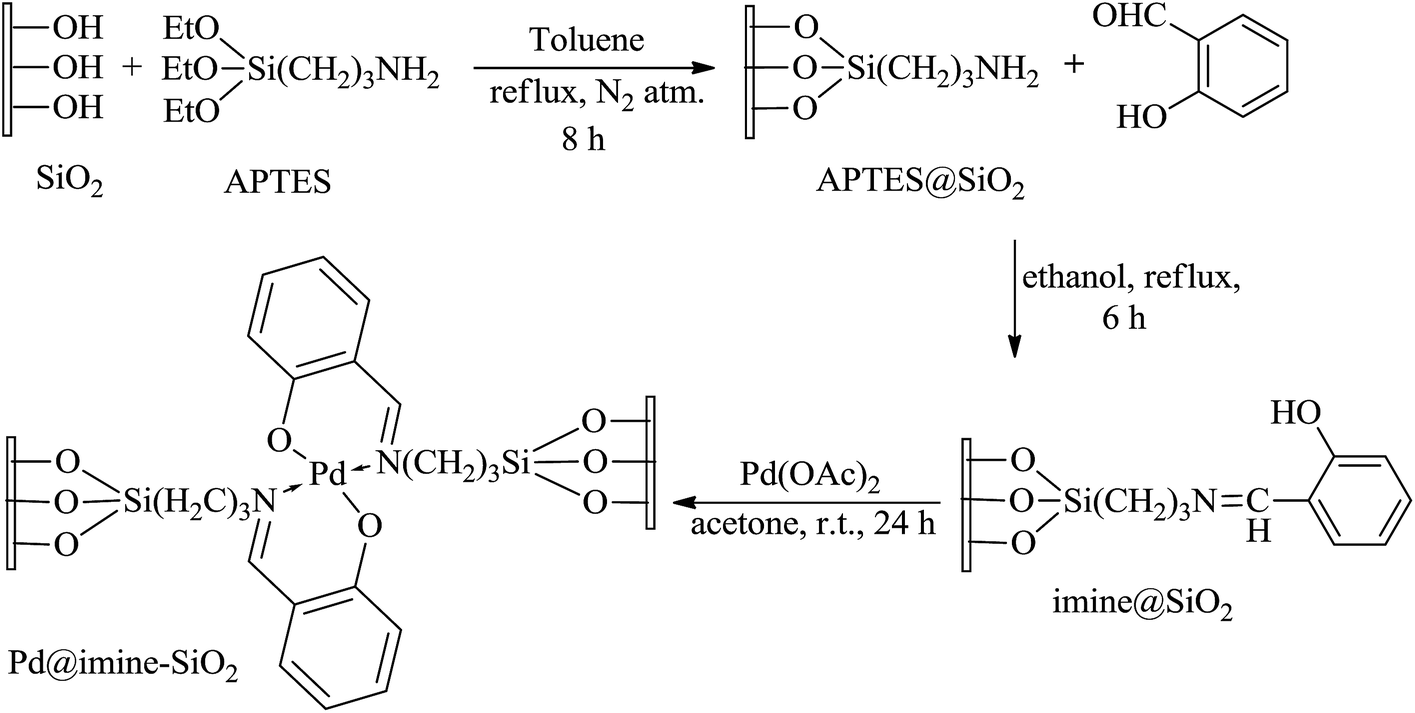 | ||
| Scheme 1 Preparation of the supported Schiff base Pd(II) complex. | ||
The first step involves the modification of silica surface by treating APTES and solid silica gel (60–120 mesh) in dry toluene under N2 atmosphere at reflux condition. In the second step, a Schiff-base ligand was synthesized by adding APTES@SiO2 and salicylaldehyde in ethanol at reflux to give the corresponding imine@SiO2 ligand, which is yellow colored solid after final purification. Finally the ligand precursor was used for the synthesis of Pd@imine–SiO2 complex by treating with Pd(OAc)2 in acetone at room temperature for 24 h.
Due to insolubility of the silica supported Pd(II) complex in almost all organic solvents, its structural investigations were limited only to its physicochemical properties, such as FTIR, SEM-EDX, EDS-mapping, ICP-AES, N2 adsorption–desorption spectral data. The FTIR spectra of SiO2, APTES@SiO2, imine@SiO2 and Pd@imine–SiO2 are presented in Fig. 1. The FTIR spectra of the samples shows absorption bands around 3400 and 1638 cm−1 for νO–H stretching and bending vibrations of the adsorbed water in the samples. The absorption peaks at 1050–1150 cm−1 are due to the stretching vibrations of the framework and Si–O–Si groups. The presence of several bands with medium intensity around 2830–2930 cm−1 region is ascribed to νC–H stretching of methylene groups. Apart from the case of I, the νN–H in the region 1500–1600 cm−1 were visible in all samples (Fig. 1). This indicates the attachments of APTES onto the mesoporous silica (I) matrix. The bands observed at 1670 cm−1 and 1460 cm−1 for III could be attributed to the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and νC–O stretching frequency, and at 540 cm−1 in IV indicates νPd–N stretching frequency.24
N and νC–O stretching frequency, and at 540 cm−1 in IV indicates νPd–N stretching frequency.24
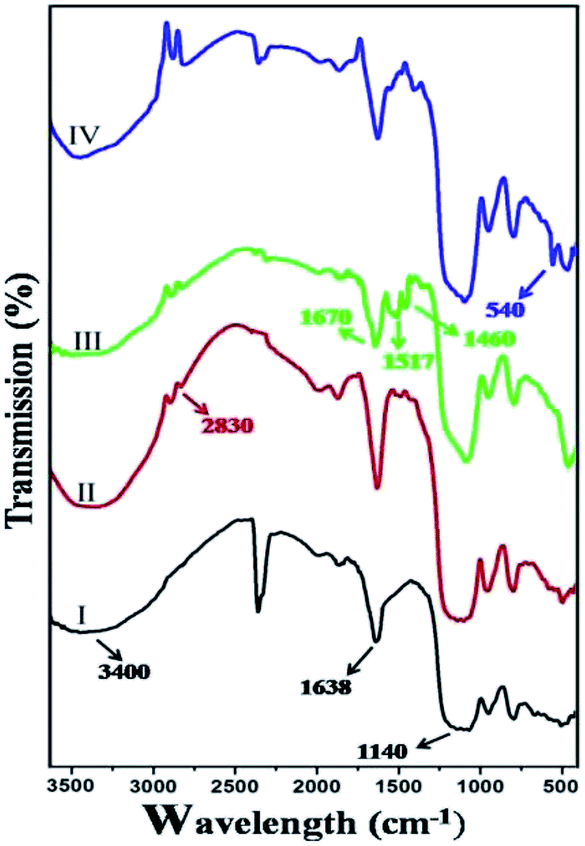 | ||
| Fig. 1 FTIR spectra; (I) silica gel (SiO2), (II) APTES@SiO2, (III) imine@SiO2 and (IV) Pd@imine–SiO2. | ||
XRD, ICP-AES, SEM-EDX and EDS mapping analysis
X-ray diffraction patterns of amorphous silica and palladium immobilized silica gel are shown in Fig. 2. Free SiO2 exhibits a peak at 2θ value of 21.10. On immobilization of palladium acetate, two characteristic peaks of Pd at 2θ = 38.12 and 47.23, corresponding to the (111) and (200) lattice planes of the face centered cubic phase of palladium were observed, which is consistent with reported literature.23,25 Considering the results of the ICP-AES and SEM-EDX observations, we were able to determine the metal attachment on the surface of solid supports. The metal content of Pd@imine–SiO2 was determined by ICP-AES, which suggests 2.465 wt% or 0.023 mmol palladium loading in the immobilized complex.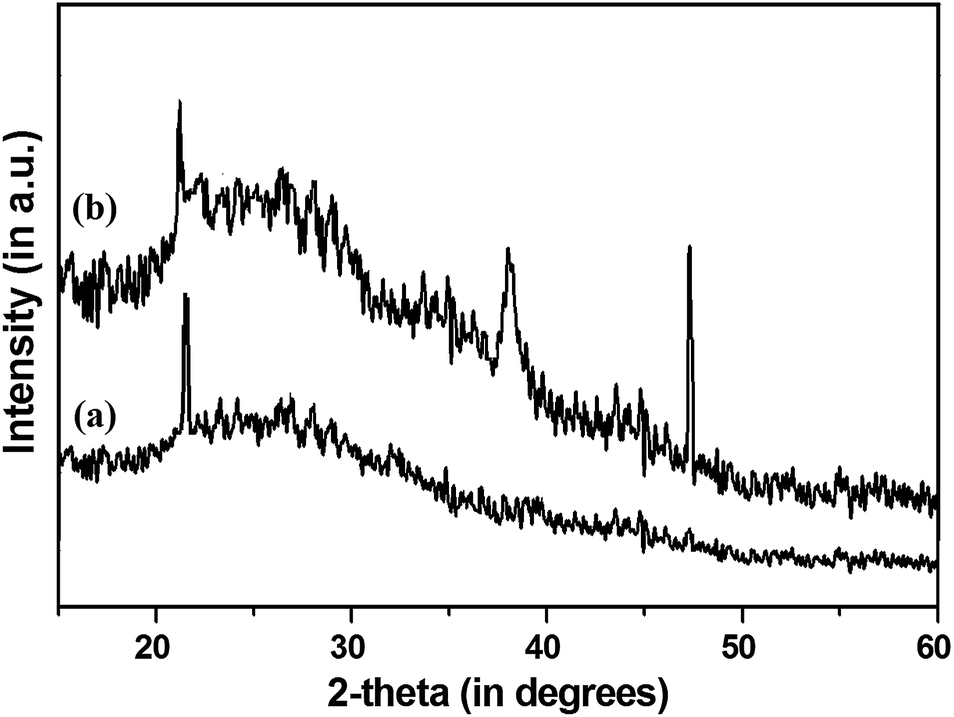 | ||
| Fig. 2 XRD pattern of (a) free silica, and (b) Pd@imine–SiO2. | ||
According to the results of the SEM-EDX spectra shown in Fig. 3, the metal complex shows metal content along with O- and Si-proportions, which confirms the formation of metal complex with the anchored ligand at various sites. The SEM image of the free SiO2 and supported Pd-catalyst clearly showed the morphological change which occurred on the surface of silica after being loaded with palladium. As intensely, observed in Fig. 4 that, the presence of palladium causes a significant decrease in the silica particle size.
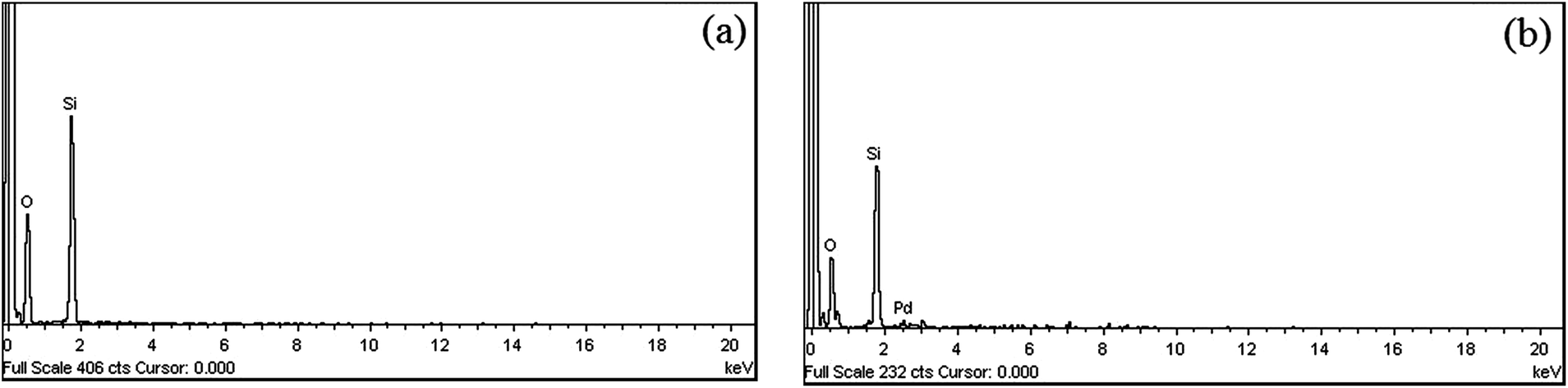 | ||
| Fig. 3 SEM-EDX spectra of (a) silica gel (SiO2) (b) Pd@imine–SiO2. | ||
 | ||
| Fig. 4 SEM image of (a) silica gel (SiO2), (b) Pd@imine–SiO2. | ||
To further confirm this Pd@imine–SiO2 structure, we take the elemental mapping by energy dispersive X-ray absorption spectroscopy (EDS). Fig. 5 shows that the distribution of active palladium sites and silica is very uniform throughout the whole matrix.
 | ||
| Fig. 5 (a) SEM image; EDS mapping of elements: (b) oxygen, (c) Si, (d) Pd and (e) Si + O + Pd. | ||
N2 adsorption–desorption isotherm
N2 adsorption–desorption isotherm analysis provides information on the specific surface area and porosity of the prepared samples. All of them exhibited the type IV isotherm according to the IUPAC classification with a typical hysteresis loop, featuring mesoporous material with highly uniform pore size distribution. Fig. 6 displays the N2 adsorption–desorption isotherm and pore size distribution of the silica based samples under study. The structural parameters are summarized in Table 1. According to BET measurement, the surface area for SiO2 was 421.9 m2 g−1 which upon functionalized with APTES and then treatment of salicylaldehyde changed from 333.2 m2 g−1 to 293.5 m2 g−1 respectively with corresponding pore volume 0.449 cm3 g−1, 0.415 cm3 g−1 and 0.374 cm3 g−1 respectively. While a decrease in the surface area (210.1 m2 g−1) and pore volume (0.274 cm3 g−1) was observed when palladium was immobilized onto imine@SiO2.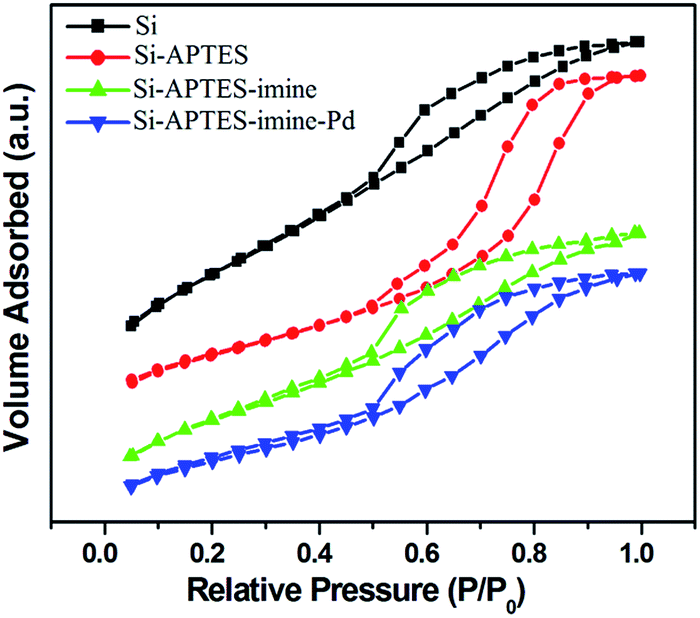 | ||
| Fig. 6 N2 adsorption–desorption isotherm of the prepared samples. | ||
| Entry | Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore radius (nm) |
|---|---|---|---|---|
| 1 | SiO2 | 421.9 | 0.449 | 2.05 |
| 2 | APTES@SiO2 | 333.2 | 0.415 | 2.20 |
| 3 | Imine@SiO2 | 293.5 | 0.374 | 2.29 |
| 4 | Pd@imine–SiO2 | 210.1 | 0.274 | 3.26 |
We also observed an increase in pore diameter in the case of Pd@imine–SiO2 (3.26 nm) than the parent SiO2 (2.05 nm). A considerable decrease in the BET surface area and pore volume compared to the solid supported SiO2 suggests the possibility of the organic moieties anchored successfully on the surface of SiO2.
Pd@imine–SiO2 catalyzed Suzuki–Miyaura reaction
Catalyst screening and base-solvent optimization
To optimize the reaction conditions, a series of experiments under varied conditions in terms of solvents, bases and the amount of catalyst for a model Suzuki coupling reaction was performed by reacting 4-bromonitrobenzene 1 with phenylboronic acid 2 as illustrated in Table 2. The reaction were carried out successfully at room temperature under open air without any special precautions. Screening of solvents using K2CO3 as a base showed that the reactions were preceded in both protic and aprotic solvents although significant variations in yields were noticed. Generally, the presence of water facilitates the solubility of the bases, which activates the boronic acid, resulting in the enhancement of the course of the reaction. This maybe the reason for low yield in EtOH, PEG (PEG400), iPrOH and THF (Table 2, entries 1, 3, 4, and 5). Although, these solvents are usually good in conventional Pd-catalyzed cross-coupling reactions,26 they exhibits lower efficiency under present reaction conditions. When pure water was used, the reaction gave poor results after 8 h (Table 2, entry 2). However, when water was used as a co-solvent with iPrOH or PEG400 quantitative yield was achieved (Table 2, entries 6, 7, 8 and 9). It is seen that the yield of the product was promising and only 58% yield was obtained in PEG400 in the absence of H2O (Table 2, entry 3). With the adding of H2O in PEG400, the yield increased (Table 2, entry 7) then decreased when the amount of water increases in the solvent system (Table 2, entry 8). The high yield of 85% was achieved when the volume ratio of PEG400 to H2O was 1:1 (Table 2, entry 7). Thus keeping H2O as co-solvent, our study revealed that iPrOH/H2O (1:1) combination is the best solvent system for our catalytic system.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Time (h) | Yieldb (%) |
| a Reaction conditions: 4-bromonitrobenzene (0.5 mmol), phenylboronic acid (0.6 mmol), base (1.5 mmol), solvent (4 mL), Pd@imine–SiO2 (20 mg, 0.463 mol% Pd), base (1.5 mmol) ca. 27 °C in air unless otherwise noted.b Isolated yield.c Pd@imine–SiO2 (15 mg).d Pd@imine–SiO2 (30 mg). | ||||
| 1 | Ethanol | K2CO3 | 6 | 30 |
| 2 | H2O | K2CO3 | 5 | 42 |
| 3 | PEG400 | K2CO3 | 5 | 58 |
| 4 | iPrOH | K2CO3 | 7 | 63 |
| 5 | THF | K2CO3 | 9 | 55 |
| 6 | Ethanol/H2O (1:1) |
K2CO3 | 7 | 65 |
| 7 | PEG400/H2O (1:1) |
K2CO3 | 4 | 85 |
| 8 | PEG400/H2O (1:3) |
K2CO3 | 8 | 40 |
| 9 | PEG400/H2O (3:1) |
K2CO3 | 5 | 78 |
| 10 | iPrOH/H2O (1:1) |
K2CO3 | 8 | 90 |
| 11 | iPrOH/H2O (1:1) |
Na2CO3 | 3 | 99 |
| 12 | iPrOH/H2O (1:1) |
Na3PO4·12H2O | 8 | 40 |
| 13 | iPrOH/H2O (1:1) |
NaOH | 6 | 52 |
| 14 | iPrOH/H2O (1:1) |
KOH | 6.5 | 56 |
| 15 | iPrOH/H2O (1:1) |
Et3N | 8 | 48 |
| 16 | iPrOH/H2O (1:1) |
Cs2CO3 | 8 | Trace |
| 17 | iPrOH/H2O (1:1) |
— | 24 | No reaction |
| 18 | iPrOH/H2O (1:1) |
Na2CO3 | 9 | 82c |
| 19 | iPrOH/H2O (1:1) |
Na2CO3 | 8 | 98d |
Further investigation was carried out to study the influence of the bases on the same reaction condition. We have found a dramatic effect of bases in the progress of the reaction. K2CO3 as a base gave moderate results in most of the organic solvents (Table 2, entries 1–9) and we obtained the good results under the same conditions in iPrOH/H2O (Table 2, entry 10). It was remarkable that, during this study, Na2CO3 was found to be the most effective base with 99% yield of the isolated product (Table 2, entry 11) under similar conditions. However, some inorganic bases like Na3PO4·12H2O, NaOH, KOH, Cs2O3 or organic bases such as Et3N resulted in lower yields (Table 2, entries 12–16). However, the reaction did not proceed in the absence of base (Table 2, entry 17). In our next move, we decided to study the influence of catalyst amount in the reaction. Decreasing the amount of catalyst resulted in lower yields under the same conditions (Table 2, entry 18) whereas higher amount of catalyst did not improve the yield or reaction time (Table 3, entry 19). Then, we successfully applied the optimized condition for the Suzuki–Miyaura cross-coupling reaction of structurally different aryl and heteroaryl bromide with arylboronic acid to produce the corresponding biaryls (Table 3). The aryl bromides with electron withdrawing and electron donating substituents underwent coupling reaction with arylboronic acid effectively to give products in high yields in iPrOH/H2O. The catalytic system is equally effective for electronically diverse arylboronic acids in the present case. These results are quite significant as the desired biaryls could be achieved at room temperature using iPrOH/H2O as a solvent and with relatively low catalyst loading. To study the scope limitation of the present catalyst, we have also studied the cross-coupling using chlorine based substrate, but found no conversion under present catalyst loading (Table 3, entries 21 and 22).
|
|||||
|---|---|---|---|---|---|
| Entry | X | R1 | R2 | Time (h) | Yieldb (%) |
| a Reaction conditions: aryl bromide (0.5 mmol), arylboronic acid (0.6 mmol), Pd@imine–SiO2 (20 mg, 0.463 mol% Pd), Na2CO3 (1.5 mmol), iPrOH/H2O (4 mL), ca. 27 °C in air unless otherwise noted.b Isolated yield. | |||||
| 1 | Br | NO2 | H | 3 | 99 |
| 2 | Br | NO2 | 4-F | 4 | 95 |
| 3 | Br | NO2 | 4-OMe | 5.5 | 94 |
| 4 | Br | OMe | H | 2 | 94 |
| 5 | Br | OMe | 4-F | 3 | 90 |
| 6 | Br | OMe | 4-OMe | 3.5 | 92 |
| 7 | Br | CHO | H | 5 | 98 |
| 8 | Br | CHO | 4-OMe | 3.5 | 97 |
| 9 | Br | CHO | 4-F | 5 | 95 |
| 10 | Br | COMe | H | 4 | 95 |
| 11 | Br | COMe | 4-OMe | 5 | 93 |
| 12 | Br | COMe | 4-F | 6 | 90 |
| 13 | Br | H | H | 1 | 100 |
| 14 | Br | H | 4-Me | 2.5 | 95 |
| 15 | Br | H | 4-F | 3.5 | 96 |
| 16 | Br | 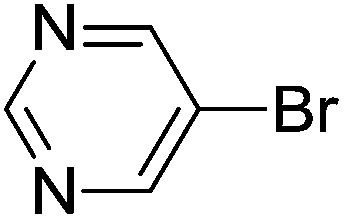 |
H | 6 | 95 |
| 17 | Br | 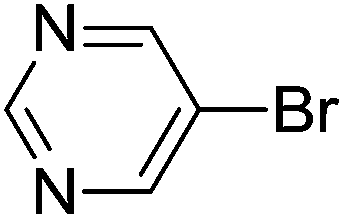 |
4-OMe | 5.5 | 96 |
| 18 | Br | 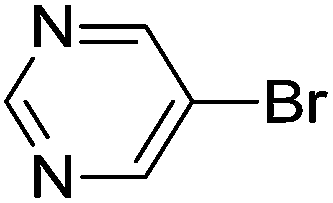 |
4-F | 7 | 97 |
| 19 | Br | OMe | 2-Me | 4 | 96 |
| 20 | Br | OMe | 3-CN | 3 | 97 |
| 21 | Cl | Me | H | 24 | No reaction |
| 22 | Cl | COMe | H | 24 | No reaction |
Since the Suzuki–Miyaura reactions are implemented on a regular basis in large scale for the preparation of pharmaceuticals, agrochemicals, polymers, natural products and advanced materials in chemical industries, we were keen to investigate the scope for scale-up to prepare grams instead of milligrams of biaryls. With the optimized reaction conditions and work-up method on small scale, we next evaluated their performance upon scale-up. Thus, the reaction of excess 4-bromonitrobenzene (5 mmol), phenylboronic acid (6 mmol), Na2CO3 (7.5 mmol), Pd@imine–SiO2 (100 mg, Pd 2.315 mol% Pd) and iPrOH/H2O (20 mL) was performed at room temperature for 3 h. To our delight, we were able to isolate 0.9885 grams (99.3%) of 4-nitro-1,1′-biphenyl.
Distinguishing between homogeneous or heterogeneous pathway
Solid catalysts are often considered to follow heterogeneous pathway, however, in certain condition they are found to function as precatalyst for more active soluble palladium species or nanoparticles. To evaluate the actual catalytic species, various control experiments have been performed.17,24 First, we pretreated 20 mg (0.463 mol% Pd) of Pd@imine–SiO2 with Na2CO3 in 50% iPrOH for 3 h and passed through a filter paper, and the filtrate was used for the coupling reaction between 4-bromonitrobenzene and phenylboronic acid. However, negligible progress (2% by GC-MS analysis) was observed upon further stirring of the catalyst-free solution for 3 h. The leached palladium in the filtrate was found to be 0.18 ppm by analysis with inductively coupled plasma atomic emission spectrometry (ICP-AES). While this result supports the strong coordination and stability of palladium ions in the catalyst which prohibits metal leaching during the reaction, it should not be considered as the sole criteria for the heterogeneity of the supported catalysts.To confirm the heterogeneity, we performed reusability test for the Pd@imine–SiO2-catalyzed Suzuki–Miyaura reaction. On the basis of our recyclability test, we found that the Pd@imine–SiO2 catalyst could be consistently used up to eight consecutive coupling reactions of phenylboronic acid with 4-bromonitrobenzene (Fig. 7). Being a solid catalyst, it could be easily recovered by centrifugation after the reaction, and after thoroughly washing with iPrOH/H2O followed by EtOAc and drying in oven, it can be used again for fresh coupling reaction. However, in successive cycle the reaction requires additional time for completion. The palladium content after 8th run was determined by ICP-AES analysis, and found to be 0.24 ppm. Since, it is confirmed that no significant leaching of palladium occurs during the catalysis and recovery, the deactivation of the catalyst after successive cycles may influence the suitable catalytic conversion (gradual decrease in yield) after successive reuse. To compare these results with conventional catalyst, we have examined the efficiency of Pd/C in the Suzuki–Miyaura reaction under identical conditions (except the catalyst). Almost negligible or trace amount of cross-couple product was obtained when the reaction of 4-bromonitrobenzene and phenylboronic acid in the presence of 0.46 mol% of 10 wt% Pd/C, Na2CO3 and isopropanol-water (1:1) was run for 3 h (compared to 99% yield with Pd@imine–SiO2 complex within 3 h).
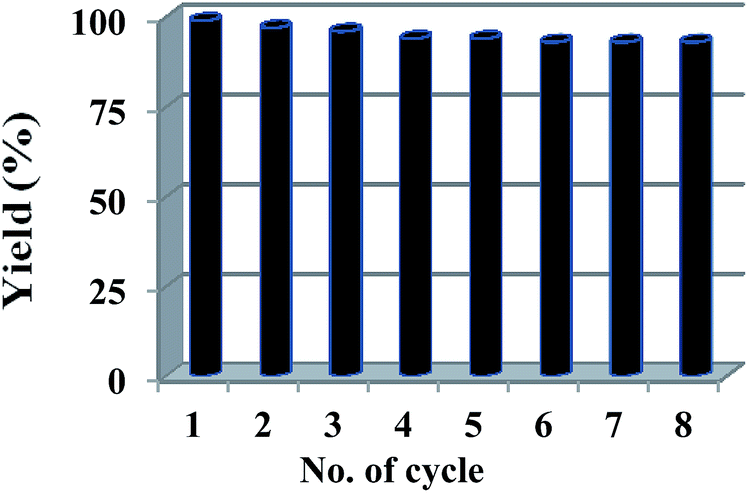 | ||
| Fig. 7 Recycling of the catalyst for the Suzuki–Miyaura reaction. | ||
Though, the negative leaching test strongly suggests that the Pd@imine–SiO2-catalyzed Suzuki–Miyaura reaction proceeds via heterogeneous mechanism, the process of “release and redepositing” back of the soluble palladium on the insoluble support during the reaction could not be ruled out.25,28 To clarify the catalytic nature of the palladium catalyst we designed catalyst poisoning test using Hg(0). Mercury is recognized as the most powerful in poisoning metals that are capable of forming an amalgam with Pt, Pd and Ni.27,29 Hg(0)-poisoning test is performed by adding excess Hg(0) to the reaction mixture prior to the addition of the catalyst. If the reaction gets suppressed by Hg(0), it evident the presence of a Pd(0) state; however, if the catalyst retains its activity, the presence of palladium in high oxidation state can be assumed. For the mercury poisoning test, Hg(0) (molar ratio to [Pd] ∼ 400) was taken in the reaction flask containing iPrOH/H2O before the addition of reactants and stirred for 0.5 h at room temperature. To this suspension 4-bromonitrobenzene (0.5 mmol), phenylboronic acid (0.6 mmol) and K2CO3 were added, and the reaction mixture was stirred for required time at room temperature. We observed that the catalyst retains its activity in the presence of large excess of Hg(0) under vigorous stirring, providing the corresponding biaryl in excellent yield without any noticeable induction time (99%, 3 h). This infers that this Suzuki–Miyaura reaction is promoted by the intact Pd@imine–SiO2 catalyst, and it is expected that the palladium remains in higher oxidation state during the reaction course.
Conclusions
In summary, we have developed a novel palladium-based heterogeneous catalyst by immobilizing Pd(OAc)2 onto imine-functionalized silica gel via coordination. The synthesized catalyst exhibited excellent activity for the Suzuki–Miyaura cross-coupling reactions of aryl bromides with arylboronic acids at low catalyst loading (20 mg, 0.463 mol% Pd). This method proceeds under aqueous media and with a recyclable catalytic system, offering an environmentally benign alternative to the existing protocols.Experimental section
General information
All chemicals were obtained commercially and used as received without further drying or purification. FT-IR spectra (4000–250 cm−1) were recorded in KBr or CHCl3 on a Shimadzu Prestige-21 FT-IR spectrophotometer. For the N2 adsorption experiments, the samples were first degassed under vacuum for 2 h at 150 °C. Subsequently, the nitrogen adsorption–desorption isotherms were determined using liq. N2 at −78 °C. The surface area of the samples was calculated according to the BET equation. Pore size distribution was evaluated using BJH algorithm. 1H spectra were recorded in CDCl3 using TMS as an internal standard on a JEOL, JNM ECS NMR spectrometer operating at 400 MHz and on Advance DPX 300 MHz FT-NMR spectrometer operating at 300 MHz. Gas chromatographic analysis coupled with mass spectrometry (GC-MS) was performed on an Agilent Technologies GC system 7820A coupled with a mass detector 5975 and SHRXI-5MS column (15 m length, 0.25 mm inner diameter, 0.25 micron film thickness). The melting points of the products were determined by using BUCHI B450 melting point apparatus. SEM analyses were carried out with JEOL, JSM Model 6390 LV scanning electron microscope, operating at an accelerating voltage of 15 kV. EDX spectra as well as element mapping analyses were also performed in the same instrument attached to the scanning electron microscope. Palladium loading in the catalyst and its possible leaching after catalytic reaction was determined using inductively coupled plasma-atomic emission spectrometry (ICP-AES) on a Thermo Electron IRIS Intrepid II XSP DUO. The course of the reaction was followed by TLC on silica gel plates (Merck, silica gel 60F254), using n-hexane-ethyl acetate as eluent. Reaction products were confirmed by comparing the 1H spectra with those reported in the literature. Silica gel with particle size 60–120 mesh was purchased from SRL Chemicals, India. 3-(Triethoxysilyl)propan-1-amine or 3-aminopropyltriethoxysilane (APTES) was purchased from Sigma Aldrich. Pd(OAc)2 was purchased from Spectrochem Pvt. Ltd., India.Catalyst preparation
APTES functionalized silica gel: modification of silica surface
The solid silica gel (60–120 mesh) was dehydrated by heating at 100 °C for 2 h prior to use. Silica (5.0 g) was refluxed with 2 mmol (442.7 mg) of APTES for 6–7 h in 50 mL of dry toluene under N2 atmosphere. After cooling, the solids were separated out by filtration and washed repeatedly through Soxhlet extraction with toluene. The solid product was dried overnight in an oven at 100 °C and the resulting material is denoted as APTES@SiO2.Synthesis of Schiff-base functionalized silica gel
The Schiff-base ligand was synthesized by adding 1.25 g of APTES@SiO2 and 1.06 mmol (129.4 mg) of salicylaldehyde in the ethanol solution and stirring for 6 h under reflux. The resulting yellow colored solid was filtered and washed repeatedly through Soxhlet extraction by ethanol followed by acetone and dried in oven at 100 °C for overnight and denoted as imine@SiO2.Immobilization of Pd(OAc)2 onto imine@SiO2
Finally, palladium derived catalyst was prepared by adding 1 g of yellowish imine@SiO2 solid with 50 mg of Pd(OAc)2 in 20 mL of acetone. The mixture was stirred at room temperature for 24 h. The solid powder became charcoal-grey in color during the stirring. The resulting solid was washed with acetone in Soxhlet and then dried. The prepared catalyst is designated as Pd@imine–SiO2 and stored for further applications.General information about catalytic experiments
Suzuki–Miyaura cross-coupling reactions were carried out under aerobic condition at room temperature (ca. 28 °C). Progress of the reactions was monitored by using aluminum-coated TLC plates (Merck silica gel 60F254) under UV lamp. The products were isolated by column chromatographic technique using silica gel (60–120 mesh). The isolated products were characterized by comparing their 1H NMR, mass spectral data and melting point with the authentic samples.Typical procedure for Suzuki–Miyaura reactions of aryl halides using palladium complex
A 50 ml round-bottom flask was charged with a mixture of aryl halide (0.5 mmol), arylboronic acid (0.6 mmol), Na2CO3 (1.5 mmol), catalyst (20 mg, Pd 0.463 mol% Pd) and solvent (4 mL). The mixture was stirred at room temperature for the required time. After completion, the reaction mixture was subjected to centrifugation and the residual solid after filtration was washed with the same solvent (4 mL) for three times. The filtrate was diluted with water (10 mL) and extracted with ether (3 × 10 mL). The resultant organic phases was washed with brine (2 × 10 ml) and dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (ethyl acetate–hexane, 1:9) to obtain the desired products. For recycling experiments, residue catalyst after filtration was washed with excess water (3 × 5 ml) and diethyl ether (3 × 5 ml) in sequence. After drying the recovered catalyst at 100 °C overnight in an oven, the residual catalyst was subjected to consequent run of the cross-coupling by charging with the required amount of substrates (aryl halide, arylboronic acid), Na2CO3 and isopropanol–water, without further addition of catalyst. The isolated products were confirmed by comparing their 1H NMR, mass spectral data and melting point with that of reported samples.
Representative example of a Suzuki–Miyaura coupling on a 5 mmol scale: reaction between 4-bromonitrobenzene and phenylboronic acid
A 100 ml round-bottom flask was charged with a mixture of 4-bromonitrobenzene (5 mmol), phenylboronic acid (6 mmol), Na2CO3 (7.5 mmol), Pd@imine–SiO2 (100 mg, Pd 2.315 mol% Pd) and iPrOH/H2O (20 mL). The mixture was stirred at room temperature for 3 h. The completion of the reaction was monitored by using TLC. After completion, the reaction mixture was subjected to centrifugation and the filtrate was diluted with water (25 mL) and extracted with ether (3 × 25 mL). The resultant organic phase was washed with brine (3 × 25 ml) and dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (ethyl acetate–hexane, 1:9) to obtain the desired products. The analytically pure product 4-nitro-1,1′-biphenyl was obtained in 0.9885 grams (99.3%).
Acknowledgements
We gratefully thank UGC, New Delhi (India) for financial support (no. 41-254/2012 (SR)). The authors are also thankful to UGC, New Delhi for Special Assistance Program (UGC-SAP) and the DST, New Delhi, for financial assistance under the DST-FIST program to the Department of Chemistry, Dibrugarh University. T. Begum and M. Mondal are thankful to UGC, New Delhi for financial assistance in the form of UGC-BSR (RFSMS) Fellowship. SAIF (STIC), Cochin is acknowledged for ICP-AES analytical service.Notes and references
- (a) J. Hassan, M. Sevignon, C. Gozzi, E. Schultz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS PubMed; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (c) X. F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047–9050 CrossRef CAS PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS; (c) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS; (d) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed; (e) A. Suzuki and Y. Yamamoto, Chem. Lett., 2011, 40, 894–901 CrossRef CAS; (f) A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181–5203 RSC; (g) A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC; (h) J. D. Sellars and P. G. Steel, Chem. Soc. Rev., 2011, 40, 5170–5180 RSC; (i) M. Mondal and U. Bora, Green Chem., 2012, 14, 1873–1876 RSC.
- (a) C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694–711 RSC; (b) F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 15, 2419–2440 Search PubMed.
- J. Zhou, X. Guo, C. Tu, X. Li and H. J. Sun, Organomet. Chem., 2009, 694, 697–702 CrossRef CAS PubMed.
- (a) W. A. Herrmann, C. P. Reisinger and M. J. Spiegler, Organomet. Chem., 1998, 557, 93–96 CrossRef CAS; (b) O. Navarro, H. Kaur, P. Mahjoor and S. P. Nolan, J. Org. Chem., 2004, 69, 3173–3180 CrossRef CAS PubMed; (c) Z. Xiong, N. Wang, M. Dai, A. Li, J. Chen and Z. Yang, Org. Lett., 2004, 6, 3337–3340 CrossRef CAS PubMed; (d) N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- (a) B. Tao and D. W. Boykin, J. Org. Chem., 2004, 69, 4330–4335 CrossRef CAS PubMed; (b) J. H. Li and W. J. Liu, Org. Lett., 2004, 6, 2809–2811 CrossRef CAS PubMed.
- D. P. Costa and S. M. Nobre, Tetrahedron Lett., 2013, 54, 4582–4584 CrossRef PubMed.
- E. Amadio, A. Scrivanti, V. Beghetto, M. Bertoldini, M. M. Alam and U. Matteoli, RSC Adv., 2013, 3, 21636–21640 RSC.
- (a) B. Banik, A. Tairai, N. Shahnaz and P. Das, Tetrahedron Lett., 2012, 53, 5627–5630 CrossRef CAS PubMed; (b) A. Dewan, U. Bora and G. Borah, Tetrahedron Lett., 2014, 55, 1689–1692 CrossRef CAS PubMed.
- A. Dewan, Z. Buragohain, M. Mondal, G. Sarmah, G. Borah and U. Bora, Appl. Organomet. Chem., 2014, 28, 230–233 CrossRef CAS PubMed.
- M. Mondal and U. Bora, Tetrahedron Lett., 2014, 55, 3038–3040 CrossRef CAS PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- (a) D. Barbaras, J. Brozio, I. Johannsen and T. Allmendinger, Org. Process Res. Dev., 2009, 13, 1068–1079 CrossRef CAS; (b) M. J. Girgis, L. E. Kuczynski, S. M. Berberena, C. A. Boyd, P. L. Kubinski, M. L. Scherholz, D. E. Drinkwater, X. Shen, S. Babiak and B. G. Lefebvre, Org. Process Res. Dev., 2008, 12, 1209–1217 CrossRef CAS; (c) J. Brown, A. Chighine, M. A. Colucci, N. Galaffu, S. C. Hirst, H. M. Seymour, J. J. Shiers, R. D. Wilkes, J. G. Williams and J. Wilson, Tetrahedron Lett., 2008, 49, 4968–4971 CrossRef CAS PubMed.
- V. Polshettiwar, C. Len and A. Fihri, Coord. Chem. Rev., 2009, 253, 2599–2626 CrossRef CAS PubMed.
- (a) H. Zhao, J. Peng, R. Xiao and M. Cai, J. Mol. Catal. A: Chem., 2011, 337, 56–60 CrossRef CAS PubMed; (b) S. Jana, S. Haldar and S. Koner, Tetrahedron Lett., 2009, 50, 4820–4823 CrossRef CAS PubMed.
- Q. Yang, S. Ma, J. Li, F. Xiao and H. Xiong, Chem. Commun., 2006, 2495–2497 RSC.
- F. Liu, G. Feng, M. Lin, C. Wang, B. Hu and C. Qi, J. Colloid Interface Sci., 2014, 435, 83–90 CrossRef CAS PubMed.
- (a) C. M. Crudden, M. Sateesh and R. Lewis, J. Am. Chem. Soc., 2005, 127, 10045–10050 CrossRef CAS PubMed; (b) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed; (c) W. Chen, P. Li and L. Wang, Tetrahedron, 2011, 67, 318–325 CrossRef CAS PubMed.
- (a) P. Veerakumar, M. Velayudham, K.-L. Lu and S. Rajagopal, Appl. Catal., A, 2013, 455, 247–260 CrossRef CAS PubMed; (b) A. Martínez, J. L. Krinsky, I. Peñafiel, S. Castillón, K. Loponov, A. Lapkin, C. Godard and C. Claver, Catal. Sci. Technol., 2015, 5, 310–319 RSC.
- (a) N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS PubMed; (b) A. Molnar, Chem. Rev., 2011, 111, 2251–2320 CrossRef CAS PubMed; (c) N. Fukaya, M. Ueda, S. Onozawa, K. K. Bando, T. Miyaji, Y. Takagi, T. Sakakura and H. Yasuda, J. Mol. Catal. A: Chem., 2011, 342–343, 58–66 CrossRef CAS PubMed; (d) S. Paul and J. H. Clark, Green Chem., 2003, 5, 635–638 RSC.
- (a) J. P. Collman, V. J. Lee, C. J. Kellen-Yuen, X. Zhang, J. A. Ibers and J. I. Brauman, J. Am. Chem. Soc., 1995, 117, 692–703 CrossRef CAS; (b) K. J. Ballus Jr, A. K. Khanmamedova, K. M. Dixon and F. Bedioui, Appl. Catal., A, 1996, 143, 159–173 CrossRef; (c) C. Heinrichs and W. F. Holderich, Catal. Lett., 1999, 58, 75–80 CrossRef CAS.
- (a) P. Wang, H. Liu, J. Niu, R. Li and J. Ma, Catal. Sci. Technol., 2014, 4, 1333–1339 RSC; (b) W. R. Reynolds, P. Plucinskibc and C. G. Frost, Catal. Sci. Technol., 2014, 4, 948–954 RSC.
- C. Sarmah, D. Sahu and P. Das, Catal. Today, 2012, 198, 197–203 CrossRef CAS PubMed.
- K. Dhara, K. Sarkar, D. Srimani, S. K. Saha, P. Chattopadhyay and A. Bhaumik, Dalton Trans., 2010, 39, 6395–6402 RSC.
- (a) A. R. Hajipour, Z. Shirdashtzade and G. Azizi, J. Chem. Sci., 2014, 126, 85–93 CrossRef CAS; (b) X. Tong, Y. Zhao, T. Huang, H. Liu and K. Y. Liew, Appl. Surf. Sci., 2009, 255, 9463–9468 CrossRef CAS PubMed.
- (a) M. V. Khedkar, T. Sasaki and B. M. Bhanage, RSC Adv., 2013, 3, 7791–7797 RSC; (b) X. Xie and S. Ma, Chem. Commun., 2013, 49, 5693–5695 RSC; (c) S. G. Modha, V. P. Mehta and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 5042–5055 RSC.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- Y. Kitamura, S. Sako, A. Tsutsui, Y. Monguchi, T. Maegawa, Y. Kitade and H. Sajiki, Adv. Synth. Catal., 2010, 352, 718–730 CrossRef CAS PubMed.
- (a) G. M. Whitesides, M. Hackett, R. L. Brainard, J. P. P. M. Lavalleye, A. F. Sowinski, A. N. Izumi, S. S. Moore, D. W. Brown and E. M. Staudt, Organometallics, 1985, 4, 1819–1830 CrossRef CAS; (b) C. Paal and W. Hartmann, Chem. Ber., 1918, 51, 711–737 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01574j |
| This journal is © The Royal Society of Chemistry 2015 |