
Targeted and controlled drug delivery using a temperature and ultra-violet responsive liposome with excellent breast cancer suppressing ability†
Hua-Fei Li‡
ac,
Cong Wu‡b,
Mao Xia‡c,
He Zhaoc,
Meng-Xin Zhaoc,
Jing Houb,
Rong Lib,
Lixin Wei*a and
Li Zhang*b
aTumor Immunology and Gene Therapy Center, Eastern Hepatobiliary Surgery Hospital Affiliated to the Second Military Medical University, 225 Changhai Road, Shanghai 200433, China. E-mail: lixinwei@smmu.edu.cn
bDepartment of Pharmacy/Laboratory Diagnosis, Changhai Hospital Affiliated to the Second Military Medical University, 168 Changhai Road, Shanghai 200433, China. E-mail: Zhanglicdma@189.cn
cInternational Joint Cancer Institute, Translational Medicine Research Institute, the Second Military Medical University, 800 Xiangyin Road, Shanghai 200433, China
First published on 10th March 2015
Abstract
Drug delivery systems (DDS) with favorable serum stability, high intra-tumor accumulation and tumor specific drug release are highly desired for promoting chemotherapeutic efficacy. However, a more stable DDS means it is more difficult to release its content, and vice versa. In order to resolve this conflict, a Fab conjugated thermo-responsive liposome (FCTRL) based on 1,2-bis(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine (DC8,9PC) and P(IPPAm-co-DMAAm)-b-PLA diblock copolymer (PPDC) was developed in this study. DC8,9PC is a type of phospholipid, which can form intermolecular cross-linking within the liposomal bilayer by ultra-violet irradiation, contributing to superior serum stability in the blood vessels. PPDC is a thermo-responsive block copolymer, which demonstrates temperature controlled ON–OFF drug release with a volume phase transition temperature (VPTT) of approximately 38.5 °C. The well modified FCTRLs are of the desired particle size, drug loading content and drug release profile. The surface morphology and pharmacokinetics were also characterized. The cellular uptake and intracellular accumulation of FCTRL are significantly promoted by its proper size, temperature regulated passive and Fab navigated active targeting. With the co-operation of all the above superiorities, the FCTRL DDS demonstrates exceptional excellent tumor suppression abilities against breast cancer in both in and ex vivo experiments, which merits further investigation in the clinic.
Introduction
Chemotherapy has been widely used against many malignancies.1 The ideal goal of chemotherapy is to deliver high-efficacy drugs at the right time to the right location at the right concentration over the right length of time.2 However, this pharmacokinetic sweet spot can hardly be achieved due to the distinctly different transient states of drugs in adhesion, distribution, metabolism and excretion (ADME).2 The clinical application of a variety of anti-cancer drugs is limited due to the serious side-effects against normal tissues because of systemic administration.3–5 Moreover, cancer cells tend to develop drug resistance, especially multidrug resistance (MDR), during the prolonged course of chemotherapy, characterized by cancer progression or recurrence.2,6 Thus, developing novel strategies for adequate and specific delivery of antineoplastic agents to tumor tissues and cells is important for an enhanced chemotherapeutic index.Anti-cancer drug delivery to malignant cells in solid tumors involves three processes, (1) transport through blood circulation to tumor vessels, (2) transport across vasculature walls into surrounding tumor tissues, (3) transport through tumor interstitial space to malignant cells. These processes are determined by the physicochemical properties of drugs and the biologic properties of tumor microenvironment, some of which are distinct from normal tissues.3,7 With the development of material science and nanotechnology, nanoparticles based drug delivery systems (DDS) offer new hopes for improving the efficiency of chemotherapy with the following potential advantages: firstly, nanoparticles with desired size (e.g., 100–200 nm in diameter) could escape from the reticuloendothelial system (RES) to realize a favorable in vivo stability in the circulating blood.8 Secondly, nanomedicine could allow the encapsulated drugs to accumulate in tumor tissues via the enhanced permeability and retention (EPR) effect and minimize the systematic toxicity.9,10 Thirdly, nanoparticles decorated with various ligands, such as monoclonal antibodies (mAbs), can realize targeted delivery and thus enhance internalization of encapsulated agents into tumor cells via endocytosis.2,5,11 Finally, nanoparticles are not physically recognized as substrates by the ATP-binding cassette (ABC) efflux pumps, which are proved to be overexpressed on chemo-resistant cancer cells and can extrude toxins from cells, conferring MDR to a broad spectrum of cytotoxic agents.12,13
Up to now, two representative nanomedicines, Doxil and Abraxane, have been approved by the U.S. Food and Drug Administration (FDA).14,15 Also, many novel nanomedicine formulations based on polymeric nanoparticles, micelles, liposomes etc. have been extensively investigated recently for their effectiveness in diagnosing and treating malignancies.14,15 Currently, increasing researches are focusing on improving the stability of the DDS in blood vessels and facilitating the release of encapsulated drugs in tumor issues and cells.2,5,16,17 However, it seems that more stable of a DDS means more difficult to release its content at the vicinity of tumors, and vice versa.16–19 The contradiction between particle stability and drug release remains a key point to resolve for further improving the tumor suppressing activities.
Herein, we constructed a Fab conjugated temperature responsive liposome (FCTRL) based on 1,2-bis(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine (DC8,9PC) and P(IPPAm-co-DMAAm)-b-PLA diblock copolymer (PPDC). DC8,9PC is a type of phospholipid, which can form intermolecular cross-linking through the diacetylenic group to produce a conjugated polymer within the hydrocarbon region of the bilayer by Ultra-Violet (UV) irradiation (Fig. 1b and d).5,20 The DC8,9PC based liposomes exhibit pleasurable serum stability after UV irradiation in our previous study.5 PPDC is a thermo-responsive block copolymer. The PPDC based Fab conjugated immunomicelles demonstrated tumor specific drug release and subsequent controlled expression of in vitro and in vivo cytotoxicity against gastric cancer cells with applied temperature changes in Jun Akimoto's study.27 In the present work, the FCTRL based on the self-assembling of DC8,9PC and PPDC not only exhibits outstanding serum stability and prolonged circulation time in the circulating blood, but also demonstrates temperature controlled ON–OFF drug release with a proper volume phase transition temperature (VPTT) of approximately 38.5 °C. What's more, with the surface decoration of Trastuzumab Fab fragments, the intra-tumor accumulation and intracellular uptake of FCTRL was further improved. The in and ex vivo experimental results reveal that drug-loading FCTRL exhibits exceptional excellent tumor suppressing ability against Her-2 (human epidermal growth factor receptor-2) positive breast cancers, which merits further investigation in the clinic.
 | ||
| Fig. 1 (a) Synthesis of P(IPAAm-co-DMAAm)-b-PLA diblock copolymers (PPDC). (b) Fabrication of Fab conjugated thermoresponsive liposomes (FCTRL). (c) Size distribution of irrad and non-irrad FCTRLs. (d) Schematic diagram of irrad and non-irrad liposomes. (e) The thermosensitive behavior of the PNIPAM and FCTRLs determined by the transmittance profile of the reflected in their volume phase transition temperature (VPTT). (f) The TEM morphology of FCTRLs at T > VPTT (left and middle panel) and T < VPTT (right panel). Scale bar: left 0.5 μm, middle 200 nm, right 0.5 μm. | ||
Materials and methods
Materials
DC8,9PC and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (Mal-PEG) were purchased from Avanti Polar Lipids (Pennsylvania, USA). PPDC was synthesized as previous descriptions (Fig. 1a).27 Chemotherapeutic drug 5-fluorouracil (5-Fu) was obtained from Melonepharma CO. LTD (Dalian, China). The CdSe/ZnS core–shell type quantum dots (Qds) and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich (St. Louis, USA). Trastuzumab (trade name: Herceptin) was purchased from Roche Pharma Ltd. (Basel, Switzerland). The Alexa Fluor 488 labeled Goat anti-human secondary antibody was obtained from Life Technologies Corporation (California, USA).Fabrication of FCTRL (Fig. 1b)
PC liposomes, composed of DC8,9PC, PPDC and mal-PEG were prepared by the thin-film hydration method as previous descriptions with minor modifications.5,20 Briefly, 2.0 mg DC8,9PC, 0.2 mg mal-PEG and 0.8 mg PPDC were dissolved in 800 μl mixed solvent of chloroform and methyl alcohol with the volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After complete dissolution, the solvent was removed in a rotary evaporator under vacuum and flashed with nitrogen to obtain a thin lipid film, which was re-suspended in 2 ml of 5-Fu solution (0.5 mg ml−1 in PBS) until completely hydrated to obtain a 5-FU loaded liposome suspension. Then, the liposome suspension was serially passed through 0.8, 0.4 and finally 0.2 μm polycarbonate membranes (Avestin, Canada). Un-encapsulated 5-FU was removed by dialysis (molecule weight cut-off, MWCO: 3 kDa) at 4 °C overnight. The resulting liposome suspensions were stored at 4 °C for future usage. The empty and Qds loaded liposomes were prepared in the same way except for washing out by respectively PBS buffer or Qds solution instead. After Trastuzumab and Bovine Serum Albumin (BSA) being thiolated by dithiothreitol as described in our previous publications,5 the resulting Trastuzumab ∼ SH and BSA ∼ SH fragments were respectively conjugated onto the liposomal surface via the reaction between the ∼SH and Mal-group at 4 °C and N2 environment overnight. The pH of the reaction mixture was adjusted to 8.4 by using sodium borate buffer. The un-conjugated fragments were removed by dialysis (MWCO: 100 kDa). For UV irradiation, liposomal solutions were exposed to 20 irradiation cycles at 4 °C, with a 254 nm UV light dose of 360 mJ cm−2 per cycle using a Stratalinker-UV 1800.5,10
:1. After complete dissolution, the solvent was removed in a rotary evaporator under vacuum and flashed with nitrogen to obtain a thin lipid film, which was re-suspended in 2 ml of 5-Fu solution (0.5 mg ml−1 in PBS) until completely hydrated to obtain a 5-FU loaded liposome suspension. Then, the liposome suspension was serially passed through 0.8, 0.4 and finally 0.2 μm polycarbonate membranes (Avestin, Canada). Un-encapsulated 5-FU was removed by dialysis (molecule weight cut-off, MWCO: 3 kDa) at 4 °C overnight. The resulting liposome suspensions were stored at 4 °C for future usage. The empty and Qds loaded liposomes were prepared in the same way except for washing out by respectively PBS buffer or Qds solution instead. After Trastuzumab and Bovine Serum Albumin (BSA) being thiolated by dithiothreitol as described in our previous publications,5 the resulting Trastuzumab ∼ SH and BSA ∼ SH fragments were respectively conjugated onto the liposomal surface via the reaction between the ∼SH and Mal-group at 4 °C and N2 environment overnight. The pH of the reaction mixture was adjusted to 8.4 by using sodium borate buffer. The un-conjugated fragments were removed by dialysis (MWCO: 100 kDa). For UV irradiation, liposomal solutions were exposed to 20 irradiation cycles at 4 °C, with a 254 nm UV light dose of 360 mJ cm−2 per cycle using a Stratalinker-UV 1800.5,10
Size distribution and morphology
The liposomal size distribution was determined by ZetaSizer (Nano-ZS, Malvern Instruments, Worcestershire, UK). To prepare stained specimens for transmission electron microscopy (TEM, Hitachi, H-7000 Electron Microscope) experiments, 5 μl liposomal suspension was dropped on 200-mesh Formvar-free carbon-coated copper grids. After the water evaporating by exposing to air at room temperature (RT), the sample was inversely covered on a drop of 2% hydrodated phosphotungstate (PTA). The TEM images were obtained at 100 kV.Temperature responsive properties
UV (UV-VIS spectrometer, Cary-100, Varian) fitted with temperature and a stirring controller was used to monitor the sample absorption profile at 300 nm. The incident light passes through the center of sample cell fitted with a thermometer. The VPTT was defined at the absorption of the liposomal suspension sharply increased to 50%.Surface binding to Her-2 antigen
The FCTRL binding to surface Her-2 antigen was assessed by confocal laser scattering microscopy (CLSM). Briefly, harvested Her-2 positive MDA-MB-231 cells were placed onto poly-D-lysine (sigma-Aldrich) coated microscope slides. Samples were fixed by 4% paraformaldehyde and successively incubated with FCTRL and Alexa-Fluor 488 labeled Goat-anti-human secondary antibodies for 1 h in room temperature (RT), respectively. The green color surrounding the cells indicates the successfully binding of Fab fragments (on liposomal surface) to Her-2 antigen (on cell surface), which was observed by CLSM (Olympus FV1000) in both 2D and 3D models.Drug releasing profile
For 5-Fu releasing profile determination, a dialysis bag (MWCO: 3 kDa) containing 3 ml irrad or non-irrad liposomal 5-Fu was respectively put into a beaker with 500 ml PBS. The beaker was fixed in a water bath respectively kept at 37 and 40 °C with continues siring. At different time intervals, approximately 500 μl solution outside the dialysis bag was sampled and measured by High Performance Liquid Chromatography (HPLC) to determine the 5-Fu concentration following previous studies.21,22 The cumulative drug release was calculated by the following equation:
Serum stability evaluation by DLS
A BSA solution in dulbecco's modified eagle medium (DMEM, GIBCO, Life Technologies, California, USA) with a concentration of 50% (m/v) was used as an in vitro serum model to mimic the in vivo status. Then the irrad and non-irrad liposomes were separately mixed with the serum model at 37 °C for 24 h. The dynamic light scattering (DLS) was used to measure the size distribution profile of BSA–liposome mixture after different time intervals, respectively.Cell culture
Two Her-2 positive human B breast cancer cell lines, MDA-MB-231 and MCF-7, were obtained from the American Type Culture Collection (ATCC). Cells were propagated and maintained in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS, GIBCO, Life Technologies, California, USA) in a controlled atmosphere incubator at 37 °C with 5% CO2.Biocompatibility evaluation
MDA-MB-231 cells (3.0 × 103) were seeded into a 96-well microplate and cultured overnight. Then cells were incubated with empty FCTRLs and BCTRLs (BSA conjugated temperature responsive liposomes) over a range of concentrations from 1 to 64 μg ml−1 in DMEM/FBS at 37 °C/40 °C. After 48 h, samples were washed and incubated with 10% Cell Counting Kit-8 (CCK-8, Beyotime Institute of Biotechnology, Shanghai, China) for 2 h protected from light. The absorbance (Ab) at 450 nm was recorded by a micro-plate reader (Thermo Multiskan MK3, Thermo Scientific) and cell viability was calculated as the following equation:where Absample represents absorbance of test well, Abcontrol represents absorbance of positive growth control well (incubated without any additional substances at each temperature), and Abblank represents absorbance of only CCK-8.
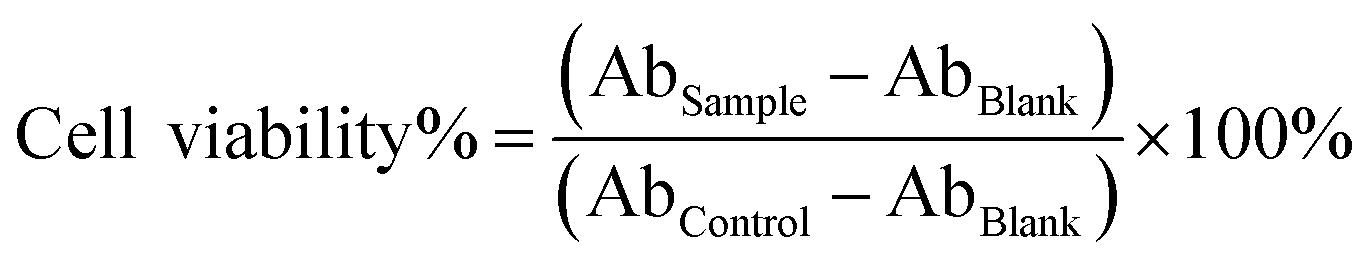
Cellular uptake and internalization
Cells were seeded into a 48-well microplate (1 × 105 cells) and incubated with 1 μg ml−1 free and liposomal Qds in DMEM/FBS at 37 and 40 °C for 4 h. Cells incubated with culture medium were used as a negative control. After washing, a FACScan Flow Cytometer (Becton Dickinson, San Jose, CA) was used to assess the cellular uptake of Qds by detecting the mean fluorescence intensity (MFI) of FL-2. Additionally, each sample was also visualized using an inverse fluorescent microscope.Animals
Four-week-old female BAL B/C nude mice and ICR mice were purchased from Shanghai Experimental Animal Center of Chinese Academic of Sciences (Shanghai, China), housed in specific pathogen-free conditions and treated in accordance with guidelines of the Committee on Animals of the Second Military Medical University (Shanghai China).Pharmacokinetics and in vivo distribution analysis
The pharmacokinetics (PKs) and in vivo distribution analysis was done following Joseph M. Tuscano's study with minor revisions.23 Briefly, 8 week-old ICR mice were randomly administrated tail vein injection of free or liposomal FITC at a dosage of 5 mg kg−1. Then 10 μl of blood were collected through tail vein nicking from each mouse at different time intervals. Samples were immediately diluted into 250 μl of 0.5 mmol L−1 EDTA-PBS, followed by a centrifugation (300 g × 5 min). Plasma FITC was extracted by acidified isopropanol (75 mmol L−1 hydrochloric acid in 90% isopropanol) at 4 °C for 20 h. The FITC concentrations were determined by measuring the fluorescence intensity at 488/520 nm (excitation/emission) and expressed as μg ml−1 (FITC/blood plasma). The data were analyzed by the PK solver software.24 For biodistribution assays, tumor bearing mice were randomly administrated tail vein injection of free and liposomal FITC at a dosage of 5 mg kg−1 (n = 3 mice per treatment). Mice were sacrificed 24 h after treatment and part of tumor, heart, liver, spleen and kidneys were removed, washed, weighed, and single-cell suspensions were made. FITC was extracted from cells and the concentrations (expressed as μg per g per tissue) were determined as described above. What's more, part of the tumor tissues were collected and subjected to frozen sections, which were detected by a CLSM.In vivo anti-tumor activity assessment
MDA-MB-231 and MCF-7 cells (1 × 107) in 100 μl of PBS buffer were inoculated subcutaneously into the lateral flank of 6 week-old BAL B/C nude mice. When tumors reached about 50–60 mm3 in volume, the inoculated mice were randomly assigned to 4 groups with 4 each for the treatment of PBS, free and liposomal (FCTRL and BCTRL) 5-Fu (5 mg kg−1) via the tail vein weekly for 3 times. Post-operation monitoring was exercised at least once a day and the tumor size was measured in two perpendicular diameters with precision calipers and calculated in a range of 60 days. The tumor volume was measured according to the following equation:| Tumor volume = Length × Width2/2 (ref. 25) |
Statistical analysis
Statistical analysis was performed by Student's unpaired t test or one-way ANOVA to identify significant differences unless otherwise indicated. Differences were considered significant at a p value of less than 0.05.Results and discussion
Size distribution and morphology
Fig. 1c indicates that a ∼32% decrease of liposomal radius was occurred after UV irradiation (from approximately 138.81 nm of non-irrad FCTRL to 94.39 nm of irrad FCTRL). We ascribe this interesting physical change to the inter-molecular cross-link of DC8,9PC in the liposomal bilayers as a result of UV irradiation polymerizing (Fig. 1d), which was validated in our and other's previous studies.5,20 The TEM results (Fig. 1f) suggest that FCTRL demonstrates a regular spherical core–shell morphology at 37 °C (T < VPTT). The well modified core–shell structure and proper size distribution make our immunoliposome an ideal drug delivery system for chemotherapeutic agents.Thermo-responsive property
Poly(N-isopropylacrylamide) (PNIPAM) is a well-known thermoresponsive polymer with a lower critical solution temperature (LCST) around 32 °C, which can be regulated by hydrophilic or hydrophobic modification.26 It should be mentioned that the UV was always used to detect the transmittance of PNIPAM based copolymer solution, in such case, the temperature at the phase transition should be named as volume phase transition temperature (VPTT) instead of LCST. Besides, surface functional groups may have remarkable influence on the VPTT of nanoparticles.26It is well established that the high metabolism of tumor cells requires much more oxygen, nutrients, gas exchange and waste removal, while the heterogeneous structure and distribution of the tumor blood vessels slows down the energy exchanging between intra- and extra-tumor. All these result in relatively higher temperature in tumor tissues than that in normal tissues.27–29 Thus, the most suitable VPTT of an ideal drug delivery system might be approximately 38–39 °C, which lies between normal body temperature (36–37 °C) and tumor temperature (approximately 40 °C). In order to tune the liposomal VPTT to this suitable temperature, FCTRLs were prepared by mixing DC8,9PC, mal-PEG and PPDC at a mass ratio of 2:0.2:0.8 based on our preliminary experimental results (data not shown). The liposomal thermoresponsive behavior in PBS was investigated with the results showing in Fig. 1e. As we can see, the FCTRL exhibits a VPTT of approximately 38.5 °C. The results were confirmed by TEM (Fig. 1f), of which the results revealed that the well modified core–shell structure of FCTRL was disrupted by increasing the local temperature to 40 °C (T > VPTT). It is helpful to know that the thermos-responsive segments (PID chain) will change from hydrophilic to hydrophobic as T > VPTT. This hydrophilic/hydrophobic transition enhanced the hydrophobicity of the block copolymer on liposomal surface. Such hydrophobicity increase leaded to the inter-liposome aggregation and/or the disruption of liposomal compartment (Fig. 1f, and ESI Fig. 2†),30,31 resulting in temperature controlled ON–OFF drug release which we will discuss later (Fig. 2b).
 | ||
| Fig. 2 (a) Confirmation of Fab conjugation on liposomal surface by CLSM. The Her-2 positive MDA-MB-231 cells were successively incubated with FCTRL and Alexa-Fluor 488 labeled Goat-anti-human secondary antibodies. The green color surrounding cells was observed by CLSM in both 2D and 3D models. Scale bar: 10 μm. (b) Drug release profile of liposomal 5-Fu at 37 and 40 °C. (c) The cytotoxicity profile of the empty FCTRLs and BCTRLs incubated with Her-2 positive MDA-MB-231 cells. | ||
Fab fragments loading
The successful anchoring of Fab fragments on liposomal surface was confirmed by CLSM. After the Her-2 positive MDA-MB-231 cells were successively incubated with FCTRL and Alexa-Fluor 488 labeled Goat-anti-human secondary antibodies, the cell morphology was observed by CLSM. The green color surrounding the cells was obviously observed in both 2D and 3D models (Fig. 2a), which directly indicated the existence of Fab fragments in the liposomal DDS.Drug loading/releasing profile
It was well expected that FCTRL could be an excellent drug carrier benefits from the stable structure. For the validation of this expectation, we firstly evaluated the 5-Fu loading content (LC) of FCTRL according to the following function: . The results revealed a relative high LC of 16.2% with our immunoliposomes.
. The results revealed a relative high LC of 16.2% with our immunoliposomes.
The drug release profiles were determined in PBS buffer respectively at 37 and 40 °C with the results showing in Fig. 2b. As we can see, both the irrad and non-irrad liposomes exhibit similar and sustained release of containing drugs at 37 °C (T < VPTT) with no initial burst during the period of 48 h. Because of PC polymerization in liposomal bilayer, slower drug release was observed with the irrad liposomes than with the non-irrad ones. By contrast, relatively rapid release of containing drugs from both irrad and non-irrad liposomes was observed at 40 °C (T > VPTT), with more than 50% and approximately 80% encapsulated 5-Fu being released after 8 and 24 h, respectively. The temperature triggered drug release can be attributed to the grafting of thermos-responsive polymers (PPDC) on liposomal surface. The PPDC displays a hydrophilic head on exterior and interior surface of liposomal bilayers (left panel of ESI Fig. 2†). When the temperature increased to T > VPTT, the PID chains will change from hydrophilic to hydrophobic. Then they will interact with the lipid bilayer inducing aggregation and destabilization, resulting in release of entrapped drugs (right panel of ESI Fig. 2†).31–33
Favorable biocompatibility
As indicated in Fig. 2c, both the empty FCTRL and BCTRL demonstrate low-cytotoxicity to MDA-MB-231 cells in concentrations of up to 64 μg ml−1 at both temperatures (37 and 40 °C). It is worth mentioning that the cell viability of FCTRL incubated cells had a little decrease compared with BCTRL incubated cells, which may be related with the tumor suppression effect of Fab fragments.Serum stability
Considering the intended use (intravenous administration), the DMEM containing 50% BSA was employed as an in vitro serum model to check the liposomal serum stability following previous publications.34 The existence of BSA was employed to mimic a variety of serum proteins in the complicated environment within the blood vessels. Table 1 shows the particle size and Polydispersity index (PDI) of irrad and non-irrad liposomes after incubation with DMEM/BSA for different time intervals. As we can see, after a 24 h-incubation, the particle radius of non-irrad liposomes was altered from 130.17 ± 24.76 nm to 187.34 ± 59.82 nm, compared with that of irrad liposomes from 93.74 ± 15.33 nm to 102.55 ± 22.7. The PDI of non-irrad liposome was increased from 0.036 to 0.102, compared with that of irrad liposomes from 0.027 to 0.050. The fewer variations of size distribution and PDI suggest that the immunoliposome owns higher stability after UV irradiation, which can also be ascribed to the PC polymerization in liposomal bilayer.| Time (h) | Non-irrad | Irrad | ||
|---|---|---|---|---|
| Raduis (nm) | PDI | Radius (nm) | PDI | |
| a PDI: polydispersity index. | ||||
| 0 | 130.17 ± 24.76 | 0.036 | 93.74 ± 15.33 | 0.027 |
| 8 | 166.72 ± 40.82 | 0.060 | 97.76 ± 20.3 | 0.043 |
| 24 | 187.34 ± 59.82 | 0.102 | 102.55 ± 22.7 | 0.050 |
Intracellular uptake
Effects of temperature and Fab decoration on intracellular uptake of the liposomes were investigated by FCM and CLSM using Qds-labeled FCTRLs and BCTRLs. Fig. 3b and d demonstrate that the red fluorescence derived from free Qds incubated cells was negligible at both 37 and 40 °C. The BCTRL and FCTRL significantly enhanced the intracellular uptake of encapsulated Qds indicated by the increased MFI of FL-2 (red fluorescence). In addition, the Fab fragments decoration can significantly enhance the intracellular uptake of encapsulated Qds at the same incubation temperature, which was approximately 1.4 and 1.55 times that of BCTRL/Qds incubated MDA-MB-231 and MCF-7 cells, respectively. We further investigated the temperature-modulated intracellular uptake. As we can see from Fig. 3b and d, the fluorescence from Qds inside the cells was clearly improved above the VPTT (40 °C) for both the FCTRLs and BCTRLs. The FCM results were validated by CLSM as displayed in Fig. 3a and c. | ||
| Fig. 3 The effects of temperature and Fab on the intracellular uptake of liposomal Qds in MDA-MB-231 (a) and MCF-7 (c) cells indicated by the inverse fluorescent microscopy. Red fluorescence represents the intracellular Qds. Magnification times: 20 × 20. The effects of temperature and Fab on the intracellular uptake of liposomal Qds in MDA-MB-231 (b) and MCF-7 (d) cells indicated by FCM. Data are mead ± SD (n = 3). | ||
It should be noted here that, firstly, the liposomes showed obvious enhancement for the intracellular uptake of encapsulated agents similar to our previous studies.5,19 Secondly, the intracellular uptake was significantly enhanced by the active targeting via antibody–antigen identification and combination. More importantly, upon temperature increasing above the VPTT, the corona-forming polymer chains collapse with the dehydration of IPAAm units. Because of significant polymer conformational changes, hydrophobic interactions between the liposomes and cell membranes increase.26,35,36 Consequently, adhesion of the liposomes to the cell surfaces was promoted, followed by further enhancement of intracellular uptake.
In vitro cytotoxicity
Fig. 4 illustrates the cell viability of Her-2 positive MDA-MB-231 and MCF-7 cell lines treated with 5-Fu loading liposomes with and without Fab conjugation, which was accessed in comparison with that of free 5-Fu at the various drug concentrations and time intervals at 37 and 40 °C. As illustrated in Fig. 4a and d, both MDA-MB-231 and MCF-7 cells showed a significantly lower cell viability after the treatment of liposomal drugs than free drugs, while FCTRL/5-Fu exhibited more potent tumor suppressing activity compared with BCTRL/5-Fu in all the tested concentrations at both temperatures due to the intracellular drug accumulation enhanced by liposomes and Fab fragments. Similarly, for the liposomal drugs, the cell viability as incubated at 40 °C (T > VPTT) was lower than that at 37 °C (T < VPTT) because the temperature promoting intracellular uptaking and drug releasing. Similar results were obtained when cells were incubated with free and liposomal 5-Fu (0.525 μg ml−1) at different time intervals from 0–72 h (Fig. 4b and e). Furthermore, the half maximal (50%) inhibitory concentration (IC50) of 5-Fu was calculated to evaluate the cytotoxicity of the liposomal drug delivery systems according to the 5-Fu concentration dependence of the cell viability profile. It was shown in Fig. 4c and f that FCTRL/5-Fu demonstrated the lowest IC50 to both MDA-MB-231 (0.09 ± 0.019 μg ml−1) and MCF-7 (0.09 ± 0.025 μg ml−1) cells at 40 °C (T > VPTT), which was about 8 times lower than that of free drugs. These results further confirmed that both the temperature and Fab decoration successfully enhanced the liposomal cytotoxicity against HER-2 positive breast cancer cells. | ||
| Fig. 4 Concentration dependent cytotoxicity evaluation of free and liposomal 5-Fu in MDA-MB-231 cells (a) and MCF-7 cells (d). Time dependent cytotoxicity evaluation of free and liposomal 5-Fu in MDA-MB-231 cells (b) and MCF-7 cells (e). The IC50 to MDA-MB-231 (c) and MCF-7 (f) cells of free and liposomal 5-Fu. Data are mead ± SD (n = 3). | ||
Pharmacokinetics
After a short injection of free and liposomal 5-Fu at 5 mg kg−1 healthy to ICR mice, the plasma drug concentrations were measured by HPLC at different time intervals. The data were analyzed using the PK solver software,22,24 and the results are all fitted to a trilocular pattern.5,36 The time-concentration curve is shown in ESI Fig. 1† and the PK parameters in Table 2. As we can see, a remarkable difference in plasma pharmacokinetics was observed after the tail vein administration of free and liposomal 5-Fu. The t1/2γ (the elimination half time in the elimination phase) was relatively longer for irrad FCTRLs (34.59 ± 1.29 h) than that for non-irrad liposomes (21.63 ± 0.60 h) and free drugs (8.63 ± 3.83 h). In accordance, the clearance (CL) was significantly reduced for irrad liposomes (0.65 ± 0.07 ml h−1 versus CLnon-irrad liposomes: 1.47 ± 0.10 ml h−1, CLfree 5-Fu: 9.74 ± 1.71 ml h−1). As we can see, liposomal drugs owned a longer circulation time in the blood vessels. Also, the circulation time was further prolonged by higher serum stability as a result of inter-molecule cross-link in the liposomal bilayers, which we have discussed above.| Parameter | Unit | Free ADR | Non-irrad | Irrad |
|---|---|---|---|---|
| a t1/2: the elimination half time, CL: clearance, Cmax: the maximum observed concentration in the plasma, AUC: area under the concentration-time curve, MRT: mean residence time. | ||||
| t1/2α | h | 0.20 ± 0.01 | 0.19 ± 0.03 | 0.22 ± 0.07 |
| t1/2β | h | 0.98 ± 0.18 | 3.83 ± 0.78 | 2.24 ± 2.21 |
| t1/2γ | h | 8.63 ± 3.83 | 21.63 ± 0.60 | 34.59 ± 1.29 |
| CL | ml h−1 | 9.74 ± 1.71 | 1.47 ± 0.10 | 0.65 ± 0.07 |
| Cmax | μg ml−1 | 55.05 ± 7.98 | 57.83 ± 2.55 | 58.17 ± 5.23 |
| AUC0-t | (μg ml−1)·h | 85.49 ± 15.1 | 455.17 ± 34.85 | 787.12 ± 94.01 |
| MRT | h | 5.74 ± 1.96 | 28.03 ± 0.69 | 49.16 ± 1.87 |
In vivo distribution and tumor accumulation assays
In order for in vivo distribution and tumor accumulation assays, tumor bearing BAL B/C nude mice were injected with free and liposomal FITC via tail vein. Twenty-four hours later, tissues were harvested and the sum total FITC was extracted and measured. Fig. 5b demonstrated that there was a significant increase in tumor FITC accumulation in FCTRL/FITC treated mice compared with BCTRL/FITC (*p = 0.039) and free FITC treated ones (**p = 0.000). The heart, liver and spleen all showed less FITC accumulation with liposomal FITC injection than with free FITC treatment (*p < 0.05). There was no difference in FITC accumulation among treatments for the kidneys (p = 0.33). The displayed fluorescent image of different frozen sections (Fig. 5a) also demonstrated significantly higher green fluorescence in tumor tissues of mice treated with liposomal FITC than with free FITC. Moreover, the intra-tumor accumulation of FCTRLs was much higher than that of BCTRLs. The intermolecular cross-linking in the liposomal bilayer as a result of UV irradiation increased the in vivo stability and prolonged its circulation time in the blood vessels. This is of great importance for tumor accumulation. As the liposomal drug delivery system arriving at the tumor site, both the passive targeting via EPR effect and active targeting via antibody–antigen reaction promoted the relatively specific intratumor accumulation. Additionally, it was reported that the temperature of some solid tumor was 1.5–3 °C higher than that of normal tissue.5,36 While the VPTT of the liposomes was about 38.5 °C, which indicated that part of the liposomal surface will transfer from hydrophilic to hydrophobic in the tumor tissues and cells. This phase transition can improve intracellular uptake which we have discussed above. On the other hand, such temperature regulated passive targeting could be very useful in conjunction with local heating cancer therapy. | ||
| Fig. 5 (a) In vivo distribution profile of frozen section from MDA-MB-231 bearing BAL B/C nude mice treated with free and liposomal FITC for 24 h as visualized by CLSM, the green fluorescence represents the tumor accumulation and retention of FITC. Scale bar: 100 μm. (b) MDA-MB-231 bearing BAL B/C nude mice were treated with free and liposomal FITC, after 24 h, mice were euthanized and organs were harvested, washed, and the FITC was extracted and quantified. Data are mean ± SEM of four separate mice in each group. (c and d) In vivo anticancer therapeutic effects in MDA-MB-231 (c) and MCF-7 (d) bearing BAL B/C nude mice after the first intravenous administration of PBS, free and liposomal 5-Fu. Data are mean ± SD of 5 separate mice in each group. | ||
In vivo tumor suppression
For the evaluation of in vivo anti-tumor activities, MDA-MB-231 and MCF-7 cells were inoculated subcutaneously into the right flank of BAL B/C nude mice. When the tumors reached about 50–60 mm3 in volume, mice were randomly treated with free and liposomal 5-Fu (5 mg kg−1). The relative tumor volume was calculated and illustrated in Fig. 5c–d. As we can see, mice treated with liposomal 5-Fu demonstrate a remarkable decrease in tumor burden compared with free 5-Fu and PBS treatment as measured by tumor volume. Otherwise, FCTRL/5-Fu exhibits a more outstanding tumor suppression ability in both breast cancer cell lines comparing with BCTRL/5-Fu, with respectively 3/5 (MDA-MB-231) and 1/5 (MCF-7) mice of complete remission (CR) indicated by no measurable mass. In our opinion, this exceptional excellent in vivo anti-tumor activity is the co-operative action of the following effects: (1) enhanced intracellular uptake due to effective endocytosis based on well-defined liposomal structure and size distribution; (2) enhanced serum stability as a result of UV irradiation polymerizing can contribute to long circulation time and durable anti-tumor activity; (3) enhanced tumor accumulation and retention in vivo through dual targeting function: passive targeting through EPR effects and active targeting through antigen–antibody reaction. (4) Relative high temperature (T > VPTT) could result in phase transition of liposomal surface PPDC from hydrophilic to hydrophobic, which can increase the hydrophobic interactions between the liposomes and cell membranes. Thus, the adhesion and following intracellular uptake of the liposomes was promoted. More importantly, this phase transition can ultimately promote the release of encapsulated drugs. This temperature controlled ON–OFF drug release at the vicinity of cancer tissues or inner cancer cells can finally contribute to sufficient and efficient local drug concentration, resulting in exceptional potent tumor suppression abilities.Conclusions
In this study, we developed a drug delivery system of 5-Fu-loaded Fab conjugated thermoresponsive liposomes (denoted as FCTRL) for sustainable & targeted delivery and temperature controlled ON–OFF release of chemotherapeutic agents as a model drug for treatment of Her-2 positive breast cancer cells. The VPTT of the immunoliposome is tuned to an ideal temperature of approximately 38.5 °C, which is between the temperatures of normal and malignant tissues. The liposomal properties, including the size distribution, well modified core–shell structure, thermoresponsive characterization and favorable biocompatibility, were clearly demonstrated. The effects of UV irradiation, temperature and Fab targeting on drug loading & releasing, serum stability, intracellular uptake, pharmacokinetics, in vivo distribution and tumor accumulation of liposomal drugs were systemically investigated. It was found that the higher serum stability and longer circulation time was successfully achieved by the crosslinking in the liposomal bilayers post UV irradiation. Also, the in vitro and in vivo tumor suppressing efficacy of 5-Fu was significantly promoted by the cooperative effects of EPR effect and temperature as well as Fab moiety. All the experimental results clearly demonstrated that the contradiction between particle stability and sufficient drug release was successfully solved by using this novel thermoresponsive immunoliposomal drug delivery system, which merits further investigation for pre-clinical experiments and clinical trials.Acknowledgements
This research was supported by National Science Foundation of China (81372330, 81372312, 81201584, 81402454, 31400778); Shanghai Science and Technology Committee (12ZR1439800, 12ZR1454200, 12431900802, 14ZD1900403); Foundation of PLA (13QNT104) and Youth fund of the Second Military Medical University (2013QN14).References
- L. Rosano, R. Cianfrocca, P. Tocci, F. Spinella, V. Di Castro, V. Caprara, E. Semprucci, G. Ferrandina, P. G. Natali and A. Bagnato, Cancer Res., 2014, 74, 7453–7464 CrossRef CAS PubMed.
- R. V. Kutty, D. T. Wei Leong and S. S. Feng, Nanomedicine, 2014, 9(5), 561–564 CrossRef CAS PubMed.
- S. H. Jang, M. G. Wientjes, D. Lu and J. L. Au, Pharm. Res., 2003, 20(9), 1337–1350 CrossRef CAS.
- R. K. Jain, J. Natl. Cancer Inst., 1989, 81(8), 570–576 CrossRef CAS PubMed.
- C. Wu, H. Li, H. Zhao, W. Zhang, Y. Chen, Z. Yue, Q. Lu, Y. Wan, X. Tian and A. Deng, Nanoscale Res. Lett., 2014, 9(1), 447 CrossRef PubMed.
- J. L. Markman, A. Rekechenetskiy, E. Holler and J. Y. Ljubimova, Adv. Drug Delivery Rev., 2013, 65(13–14), 1866–1879 CrossRef CAS PubMed.
- J. L. Au, S. H. Jang, J. Zheng, C. T. Chen, S. Song, L. Hu and M. G. Wientjes, J. Controlled Release, 2001, 74(1–3), 31–46 CrossRef CAS.
- R. V. Kutty and S. S. Feng, Biomaterials, 2013, 34(38), 10160–10171 CrossRef CAS PubMed.
- H. Maeda, J. Controlled Release, 2012, 164(2), 138–144 CrossRef CAS PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11(17–18), 812–818 CrossRef CAS PubMed.
- M. R. Kano, Y. Bae, C. Iwata, Y. Morishita, M. Yashiro, M. Oka, T. Fujii, A. Komuro, K. Kiyono, M. Kaminishi, K. Hirakawa, Y. Ouchi, N. Nishiyama, K. Kataoka and K. Miyazono, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(9), 3460–3465 CrossRef CAS PubMed.
- S. C. Hyde, P. Emsley, M. J. Hartshorn, M. M. Mimmack, U. Gileadi, S. R. Pearce, M. P. Gallagher, D. R. Gill, R. E. Hubbard and C. F. Higgins, Nature, 1990, 346(6282), 362–365 CrossRef CAS PubMed.
- S. V. Ambudkar, I. W. Kim and Z. E. Sauna, Eur. J. Pharm. Sci., 2006, 27(5), 392–400 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160(2), 117–134 CrossRef CAS PubMed.
- M. W. Saif, JOP, 2013, 14(6), 686–688 Search PubMed.
- K. Ulbrich and V. Subr, Adv. Drug Delivery Rev., 2004, 56(7), 1023–1050 CrossRef CAS PubMed.
- B. Kneidl, M. Peller, G. Winter, L. H. Lindner and M. Hossann, Int. J. Nanomed., 2014, 9, 4387–4398 CAS.
- W. Li, M. Zhao, C. Ke, G. Zhang, L. Zhang, H. Li, F. Zhang, Y. Sun, J. Dai, H. Wang and Y. Guo, BioMed Res. Int., 2013, 2013, 305089 Search PubMed.
- W. Li, H. Li, J. Li, H. Wang, H. Zhao, L. Zhang, Y. Xia, Z. Ye, J. Gao, J. Dai, H. Wang and Y. Guo, Int. J. Nanomed., 2012, 7, 4661–4677 CrossRef CAS PubMed.
- C. F. Temprana, E. L. Duarte, M. C. Taira, M. T. Lamy and S. Del Valle Alonso, Langmuir, 2010, 26(12), 10084–10092 CrossRef CAS PubMed.
- J. Ciccolini, C. Mercier, M. F. Blachon, R. Favre, A. Durand and B. Lacarelle, J. Clin. Pharm. Ther., 2004, 29(4), 307–315 CrossRef CAS PubMed.
- W. R. Wrightson, S. R. Myers and S. Galandiuk, Biochem. Biophys. Res. Commun., 1995, 216(3), 808–813 CrossRef CAS PubMed.
- J. M. Tuscano, S. M. Martin, Y. Ma, W. Zamboni and R. T. O'Donnell, Clin. Cancer Res., 2010, 16(10), 2760–2768 CrossRef CAS PubMed.
- Y. Zhang, M. Huo, J. Zhou and S. Xie, Comput. Meth. Programs Biomed., 2010, 99(3), 306–314 CrossRef PubMed.
- M. Oba, K. Miyata, K. Osada, R. J. Christie, M. Sanjoh, W. Li, S. Fukushima, T. Ishii, M. R. Kano, N. Nishiyama, H. Koyama and K. Kataoka, Biomaterials, 2011, 32(2), 652–663 CrossRef CAS PubMed.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Biomacromolecules, 2009, 10(6), 1331–1336 CrossRef CAS PubMed.
- O. Tredan, C. M. Galmarini, K. Patel and I. F. Tannock, J. Natl. Cancer Inst., 2007, 99(19), 1441–1454 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100(1), 57–70 CrossRef CAS.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144(5), 646–674 CrossRef CAS PubMed.
- W. Li, J. Li, J. Gao, B. Li, Y. Xia, Y. Meng, Y. Yu, H. Chen, J. Dai, H. Wang and Y. Guo, Biomaterials, 2011, 32(15), 3832–3844 CrossRef CAS PubMed.
- B. Kneidl, M. Peller, G. Winter, L. H. Lindner and M. Hossann, Int. J. Nanomed., 2014, 9, 4387–4398 CAS.
- Y. Wu, J. Yao, J. Zhou and F. Z. Dahmani, Int. J. Nanomed., 2013, 8, 3587–3601 Search PubMed.
- M. van Elk, R. Deckers, C. Oerlemans, Y. Shi, G. Storm, T. Vermonden and W. E. Hennink, Biomacromolecules, 2014, 15(3), 1002–1009 CrossRef CAS PubMed.
- S. V. Mussi, R. Sawant, F. Perche, M. C. Oliveira, R. B. Azevedo, L. A. Ferreira and V. P. Torchilin, Pharm. Res., 2014, 31(8), 1882–1892 CrossRef CAS PubMed.
- Y. Akiyama, A. Kikuchi, M. Yamato and T. Okano, Langmuir, 2004, 20(13), 5506–5511 CrossRef CAS.
- Y. Wu, Y. Yang, F. C. Zhang, C. Wu, W. L. Lu and X. G. Mei, J. Liposome Res., 2011, 21(3), 221–228 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01553g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |