
A sensitive and selective chemiluminogenic probe for palladium†
Abstract
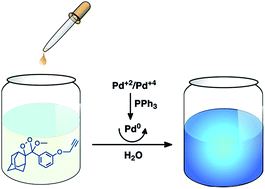
Palladium triggered removal of a propargyl group leads to the cleavage of the 1,2-dioxetane ring, leading to bright chemiluminescence. The reaction of the probe is highly specific for the Pd species, thus the probe described here has considerable potential for practical utility.
 Please wait while we load your content...
Please wait while we load your content...

