
Mesoporous SnO2 nanoparticle films as electron-transporting material in perovskite solar cells
Abstract
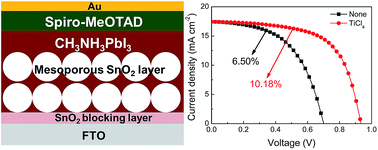
Perovskite solar cells with mesoporous metal oxide films as scaffold layers have demonstrated very impressive advances in performance recently. Here, we present an investigation into mesoporous perovskite solar cells incorporating mesoporous SnO2 nanoparticle films as electron-transporting materials and scaffold layers, to replace traditional mesoporous TiO2 films. We have optimized the SnO2 film thickness and treated the surface of the SnO2 film with an aqueous solution of TiCl4. Due to the TiCl4 treatment the recombination process was significantly retarded. The short-circuit current density (Jsc) and open-circuit voltage (Voc) reached nearly 18 mA cm−2 and 1 V, respectively. Consequently, the power conversion efficiency of the device with the SnO2 film exceeded 10%.
 Please wait while we load your content...
Please wait while we load your content...

