
Ultralong mesoporous ZnO nanowires grown via room temperature self-assembly of ZnO nanoparticles for enhanced reversible storage in lithium ion batteries†
Abstract
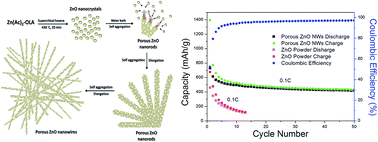
A facile template-free method for the synthesis of high purity ultralong mesoporous ZnO nanowires (up to 50 μm) has been developed. ZnO nanoparticles were first formed by decomposition of the Zn(Ac)2–OLA complex in supercritical hexane at 430 °C for 20 min. Afterward, porous ZnO nanowires were produced though in situ self-aggregation of ZnO nanoparticles at room temperature under the cooperation of oleylamine and porous metal–organic frameworks which coordinate ZnO nanoparticles aggregated together in the form of 1D porous nanostructures. Due to the porous structures, the obtained porous ZnO nanowires have a larger surface area than those of ZnO nanoparticles and ZnO nanowires which have solid structures. Furthermore, compared to the poor capacity performance of ZnO powders, giving a capacity of only around 120 mA h g−1 at 10th cycles at 0.1 C charge/discharge rate, the porous ZnO nanowire electrode shows a reversible capacity of 432 mA h g−1 after 50 cycles. The improved capacity and cycle life of the porous ZnO nanowires electrode was attributed to the structure which gives rise to more efficient Li ion transport and alleviates the large mechanical strain during lithiation and delithiation.
 Please wait while we load your content...
Please wait while we load your content...

