
Novel Bi12ZnO20–Bi2WO6 heterostructures: facile synthesis and excellent visible-light-driven photocatalytic activities†
Lei Zhangab,
Xiang Zhanga,
Yu-Qi Huanga,
Cheng-Ling Pan*ab,
Jin-Song Hu*a and
Chang-Min Houb
aLaboratory of Multiscale Materials and Molecular Catalysis, School of Materials Science and Engineering, Anhui University of Science and Technology, Huainan, Anhui 232001, P. R. China. E-mail: clpan@aust.edu.cn; jshu@aust.edu.cn
bState Key Lab of Inorganic Synthesis & Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China
First published on 23rd March 2015
Abstract
Novel sillenite-type Bi12ZnO20–Bi2WO6 heterostructure photocatalysts were successfully prepared via a controllable partial precipitate conversion strategy, employing previously-prepared Bi2WO6 nanosheets as precursors of Bi3+. The phases and morphologies of the products were characterized by powder X-ray diffraction, energy dispersive spectrometry, X-ray photoelectron spectroscopy, high resolution transmission electron microscopy and scanning electron microscopy. Compared to the sample (Bi12ZnO20 + Bi2WO6) obtained by physical mixing technology and the single-component photocatalyst (Bi12ZnO20 and Bi2WO6), such sillenite-type Bi12ZnO20–Bi2WO6 heterostructures exhibited higher visible-light-driven photocatalytic activity, which could be ascribed to matching band positions and intimate interfacial contact between different components that could benefit the efficient separation of photo-generated carriers.
1. Introduction
In the past decades, semiconductor photocatalysis has been attracting much attention due to its potential applications in the decomposition of organic waste-water, H2 production by water splitting and CO2 reduction.1–5 As is well known, a prerequisite for the development of photocatalysis applications is to gain access to efficient photocatalysts. While TiO2 can be recognized as the “star photocatalyst” owing to its high oxidizing efficiency, high chemical stability, nontoxicity and low-cost,6–8 it only responds to UV light, that accounts for about 4% of the solar radiation energy, which inevitably leads to a huge waste of energy.9 Therefore, a great deal of effort has been devoted to the development of highly efficient visible-light-driven photocatalysts.Bi12(BixM1−x)O20 (M = Zn, Si, V, Fe, Ge, Ti) belongs to a family of sillenite compounds with a body-centered cubic crystal structure (space group I23).10–12 Several years' research have shown that sillenite-type semiconductors usually possess excellent mobility of photo-induced carriers, strong oxidizing ability of photo-generated holes, and a relative narrow band-gap (<2.8 eV).13 Therefore, these kind photocatalysts should exhibit excellent visible light activity based on the theoretical analysis.14–17 Actually, many research groups have been working on the structure optimization and performance control of this kind photocatalytic material and have made encouraging progress in this field. For example, Hou and coworkers reported the successful preparation of visible-light-driven Bi12TiO20 photocatalysts with different morphology through a facile solution-phase hydrothermal process.18 Wan et al. synthesized uniform Bi12GeO20 microspheres and microtetrahedrons enclosed by four {111} facets.19 Photocatalytic experiments showed that the as-obtained samples could degrade some organic pollutants such as RhB and gaseous formaldehyde under visible light irradiation. Unfortunately, the reported photocatalytic activity of these materials is usually relatively low. The main limiting factor can be ascribable to the unfavorable recombination rate of photo-generated charges.20–26
Recently, semiconductor coupling technology has been demonstrated as an effective strategy for efficient separation of photo-generated electron–hole pairs, leading to the enhancement of photocatalytic activity. For example, some composite photocatalysts including Bi2O3–Bi2WO6,27 Bi25FeO40–graphene,28 CuO–In2O3 (ref. 29) and Bi12TiO20–TiO2 (ref. 30) all exhibit much better photocatalytic activity than the corresponding single semiconductors. In addition, among various synthesis approaches of these heterostructures, it has been demonstrated that the partial precipitate conversion method exhibits great advantages for the construction of heterostructure photocatalyst with intimate interfacial contacts between different components.31–33 This feature is particularly important for the migration, transfer and separation of photo-generated charges and the remarkable improvement of photocatalytic activity.
Therefore, we now report the successful construction of novel Bi12ZnO20–Bi2WO6 (denoted as BZO–BWO) heterostructure photocatalysts via a controllable partial precipitate conversion strategy, employing previously-prepared BWO nanosheets as a precursor of Bi3+. Significantly, our photocatalytic experiments reveal that these heterostructures exhibit excellent visible-light-driven photocatalytic activity. To the best of our knowledge, report on the preparation of such sillenite-type heterostructures is still quite rare, specially for BZO. More importantly, this work not only enriches the preparation technology and improves the photocatalytic activity of BZO, but also presents a general and effective method to improve the visible light activity of other sillenite-type photocatalytic materials.
2. Experimental section
Preparation of BWO nanosheets
The synthesis method of BWO is based on ref. 34. In a typical preparation procedure, Bi(NO3)3·5H2O (0.001 mol) was dissolved in 15 mL of deionized water. Then, 15 mL of deionized water containing 0.0005 mol of Na2WO4·2H2O was slowly dropped into the above solution. Finally, 1 g of poly(vinylpyrrolidone) were introduced. The as-obtained solution was stirred for 10 min and transferred into stainless-steel autoclave with a Teflon liner of 40 mL capability. After treating the mixture at 180 °C for 24 h, it was cooled to room temperature naturally. The product was collected, washed with deionized water and absolute ethanol, and dried in a vacuum at 60 °C for 6 h with a yield of 90%. Fig. S1† show the XRD pattern, EDS spectrum and SEM images of the product, which demonstrates the successful preparation of BWO nanosheets (see ESI†).Preparation of BZO microcrystals
In a typical preparation procedure, Bi(NO3)2·5H2O (0.0012 mol) was dissolved in 15 mL of deionized water. Then, 15 mL of deionized water containing 0.0001 mol of Zn(CH3COO)2·2H2O was slowly dropped into the above solution. Finally, 0.2 g of NaOH was introduced. The as-obtained solution was stirred for 10 min and transferred into stainless-steel autoclave with a Teflon liner of 40 mL capability. After treating the mixture at 180 °C for 24 h, it was cooled to room temperature naturally. The product was collected, washed with deionized water and absolute ethanol, and dried in a vacuum at 60 °C for 6 h with a yield of 90%. Fig. S2 and S3† show the XRD pattern, SEM images and XPS spectra of the product, which demonstrates the successful preparation of BZO microcrystals (see ESI†).Preparation of BMO–BWO heterostructures
0.5 g of BWO nanosheets, a certain amount of Zn(CH3COO)2·2H2O and 0.3 g of NaOH were introduced into Teflon liner. Then, 30 mL of deionized water was slowly dropped into the above container. The as-obtained solution was stirred for 10 min and transferred into stainless-steel autoclave. After treating the mixture at 180 °C for 24 h, it was cooled to room temperature naturally. The product was collected, washed with deionized water and absolute ethanol and dried in a vacuum at 60 °C for 6 h. The BZO–BWO heterostructures with different BZO–BWO weight ratios (9, 16 and 22%, this data is calculated from the feed ratio) were denoted as S1, S2 and S3, respectively.Characterization
The products were characterized by XRD with a Shimadzu XRD-6000 powder X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å), recorded with 2θ ranging from 15° to 70°. SEM images of the products were obtained on field emission scanning electron microanalyser (Hitachi S-4800), employing the accelerating voltage of 5 kV. Energy dispersive X-ray spectroscopy, being attached to the scanning electron microscope, was used to analyze the composition of the sample. HRTEM were carried out on a JEM-2100 high resolution transmission microscope, employing an accelerating voltage of 200 kV. XPS spectrum was recorded on a PHI5000 VersaProbe X-ray photoelectron spectrometer. The UV-vis diffusion reflectance spectrum of the sample was analyzed with a UV-vis spectrophotometer (UV-3600, Shimadzu, Japan). Photoluminescence spectrum was recorded employing an Edinburgh FLSP 920 fluorescence spectrophotometer at room temperature. Photocurrent was also measured using a CHI 660B electrochemical workstation in a standard three-electrode system using the prepared samples as the working electrodes with an active area of ca. 0.5 cm2, a platinum wire as counter electrode, and a standard calomel electrode (SCE) as reference electrode. A 300 W xenon lamp with a 400 nm cut-off filter was used as the source of visible-light irradiation. Potentials were given with reference to SCE. The photoresponses of the photocatalysts at light on and off were measured at 0.0 V.Photocatalytic evaluation
A 240 W Xe lamp (BL-GHX-Xe-300, Shanghai BILON Co., Ltd.) was used as the light source and equipped with an ultraviolet cutoff filter to provide visible light (λ ≥ 400 nm). The distance between the liquid surface of the suspension and the light source was set about 15 cm. The photocatalytic experiments were performed with the sample powder (40 mg) suspended in RhB (10 mg L−1, 40 mL) with constant stirring. Before light was turned on, the solution was continuously stirred for 60 min in the dark to ensure the establishment of an adsorption–desorption equilibrium. During irradiation, ∼3 mL of the suspension was continually taken from the reactor at given time intervals. The photocatalyst powders and the pollutions solution were separated by a centrifugal machine. The RhB concentration was analyzed through a UV-vis spectrophotometer (UV-3600, Shimadzu, Japan).3. Results and discussion
The successful construction of BZO–BWO heterostructures (S1) was confirmed by the XRD pattern (Fig. 1a). In addition to the characteristic diffraction peaks assigned to orthorhombic BWO (JCPDS file Card no. 79-2381), the remaining peaks can be well-indexed to cubic BZO (JCPDS file Card no. 78-1325). These experimental results demonstrate that the as-synthesized products are composite materials. The strong and sharp diffraction peaks suggest good crystallinity of the product and no other impurity peak is detected. The microstructure and morphology of the product were investigated by SEM. As shown in Fig. 1b, one can find that the as-prepared product is composed of cube-like microstructures and nanosheets. High magnification SEM image shown in Fig. 1c reveals that some nanosheets exist in the surface of cube-like microstructures, indicating the formation of BZO–BWO heterojunctions.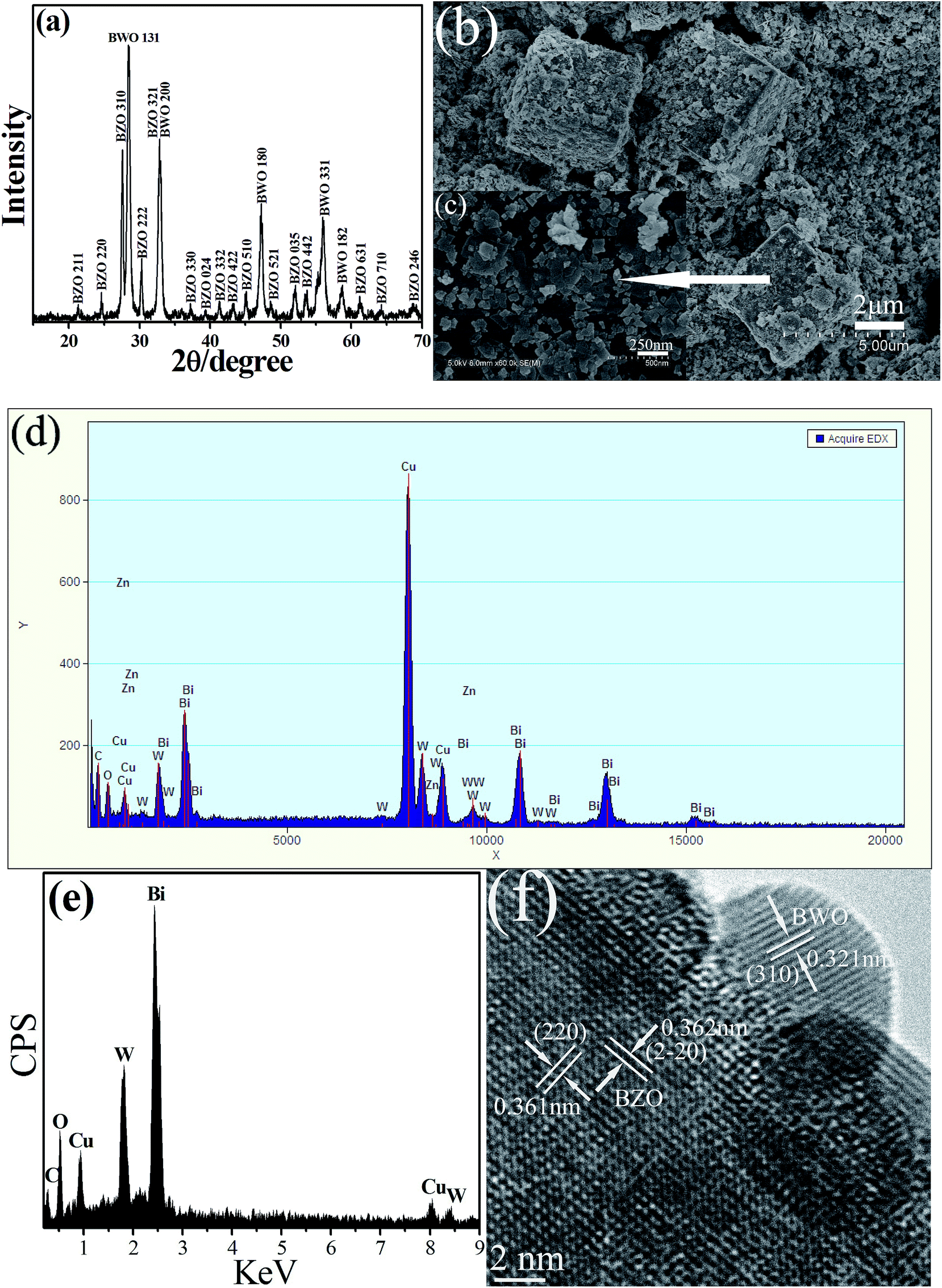 | ||
| Fig. 1 XRD pattern (a), SEM images (b and c), EDS spectra (d and e) and TEM image (f) of BZO–BWO heterostructures (S1) prepared under hydrothermal synthesis method. | ||
Because it is difficult to obtain the EDS spectrum of “pure” cube, whole heterostructure (cube and nanosheet) is selected as information acquisition area. As shown in Fig. 1d, one can find that weak Zn peak appears accompanied by peaks of other elements such as Bi, W, O. Furthermore, the EDS analysis of nanosheets attached to the surface of the cube reveals that these sheet-like structures can be assigned to BWO (Fig. 1e). Therefore, it is reasonable to believe that the remaining cube-like structure can be attributable to BZO. The further evidence of the formation of BZO–BWO heterostructures comes from HRTEM. The HRTEM image from the heterostructures displays two types of clear lattice fringes as shown in Fig. 1f. One set of the fringes spacing is ca. 0.321 nm, corresponding to the (310) plane of orthorhombic BWO. Another set of the fringes spacing measures ca. 0.361 and 0.362 nm, which corresponds to the (220) and (2–20) lattice spacing of cubic BZO, respectively. The above experimental results fully prove the formation of BZO–BWO heterojunction.
More detailed structural information regarding the chemical and bonding environment of the BZO–BWO heterostructures (S1) was ascertained using X-ray photoelectron spectroscopy. Fig. 2a shows the survey spectrum of the sample in the range of 2–1200 eV. This overview spectrum demonstrates that Bi, W and O exist in the final products. The Bi 4f peaks centered at 158.5 (Bi 4f7/2) and 163.9 eV (Bi 4f5/2) imply the existence of Bi in the Bi3+ oxidation state (Fig. 2b). Two peaks at 34.7 (W 4f7/2) and 36.7 eV (W 4f5/2) indicate that W is in its [WO4]2− (W6+) state (Fig. 2c). A strong oxygen 1s peak centered at 529.9 eV corresponds to the oxygen in its O2− oxidation state (Fig. 2d). Two weak peaks taken for the Zn region at 1021.7 eV (Zn 2p3/2) and 1044.2 eV (Zn 2p1/2) are assigned to the Zn 2p binding energy (Fig. 2e), which infers the existence of Zn in the Zn2+ oxidation state. Compared to pure BZO and BWO, the XPS peak position of Bi 4f, Zn 2p and W4f in the BZO–BWO heterostructures shift to high binding energies (Table 1),34 which may be ascribed to the weak interaction between BZO and BWO resulting from their intimate interfacial contacts.35 The weak peaks of Zn 2p can be ascribed to the low content of BZO (BZO–BWO = 9%, wt%, S1) in the final heterostructures photocatalyst.
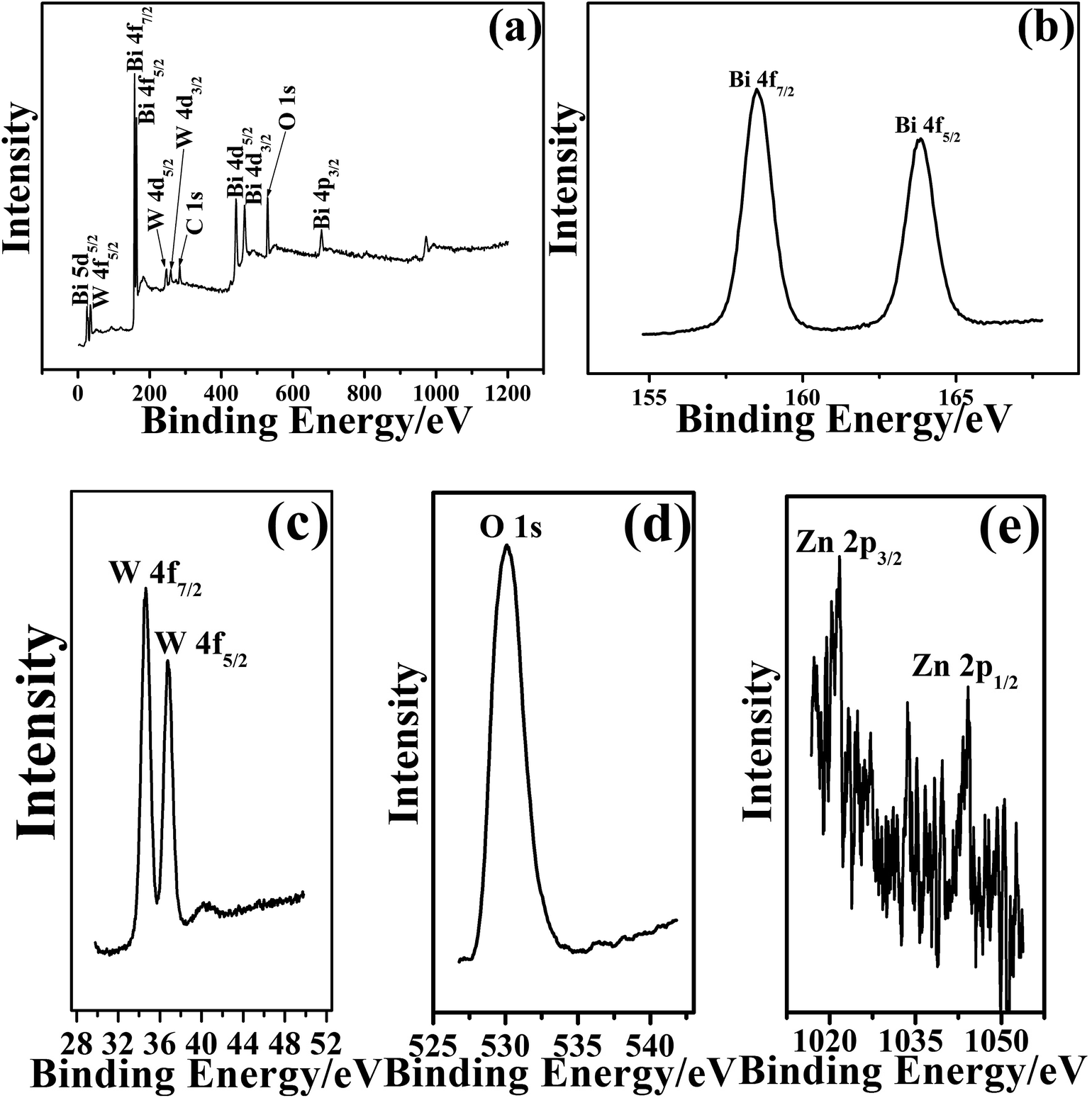 | ||
| Fig. 2 XPS spectra of BZO–BWO heterostructures (S1) prepared under hydrothermal method. | ||
| Sample | Bi 4f (eV) | Zn 2p (eV) | W 4f (eV) |
|---|---|---|---|
| S1 | 158.5, 163.9 | 1021.8, 1044.2 | 34.7, 36.8 |
| Pure BZO | 158.2, 163.6 | 1020.7, 1043.9 | — |
| Pure BWO | 158.2, 163.5 | — | 34.5, 36.5 |
If the weight percentage of BZO is increased from 9 to 30%, such XPS peak intensity of Zn 2p significantly increases but with no phase change (see ESI, Fig. S4 and S5†). For the BZO–BWO heterostructure, it is actually composed of BWO and BZO. Therefore, it should show the diffraction peaks of each component in the XRD pattern of heterostructure photocatalyst. In addition, this XRD pattern of heterostructures should also be consistent with the sample (BZO + BWO) prepared by the physical mixing method. As given in Fig. S6 (see ESI†), one can find that the sample prepared by physical mixture has almost the same XRD pattern as the BZO–BWO heterostructures. Therefore, these results further confirm the successful construction of BZO–BWO heterojunction photocatalyst with close interconnection.
The UV-vis diffuse reflection data is used to calculate the absorption coefficient from the well-known Kubelka–Munk (KM) function, defined as:
| F(R∞) = α/S = (1R∞)2/R∞ | (1) |
| αE = C(E − Eg)n | (2) |
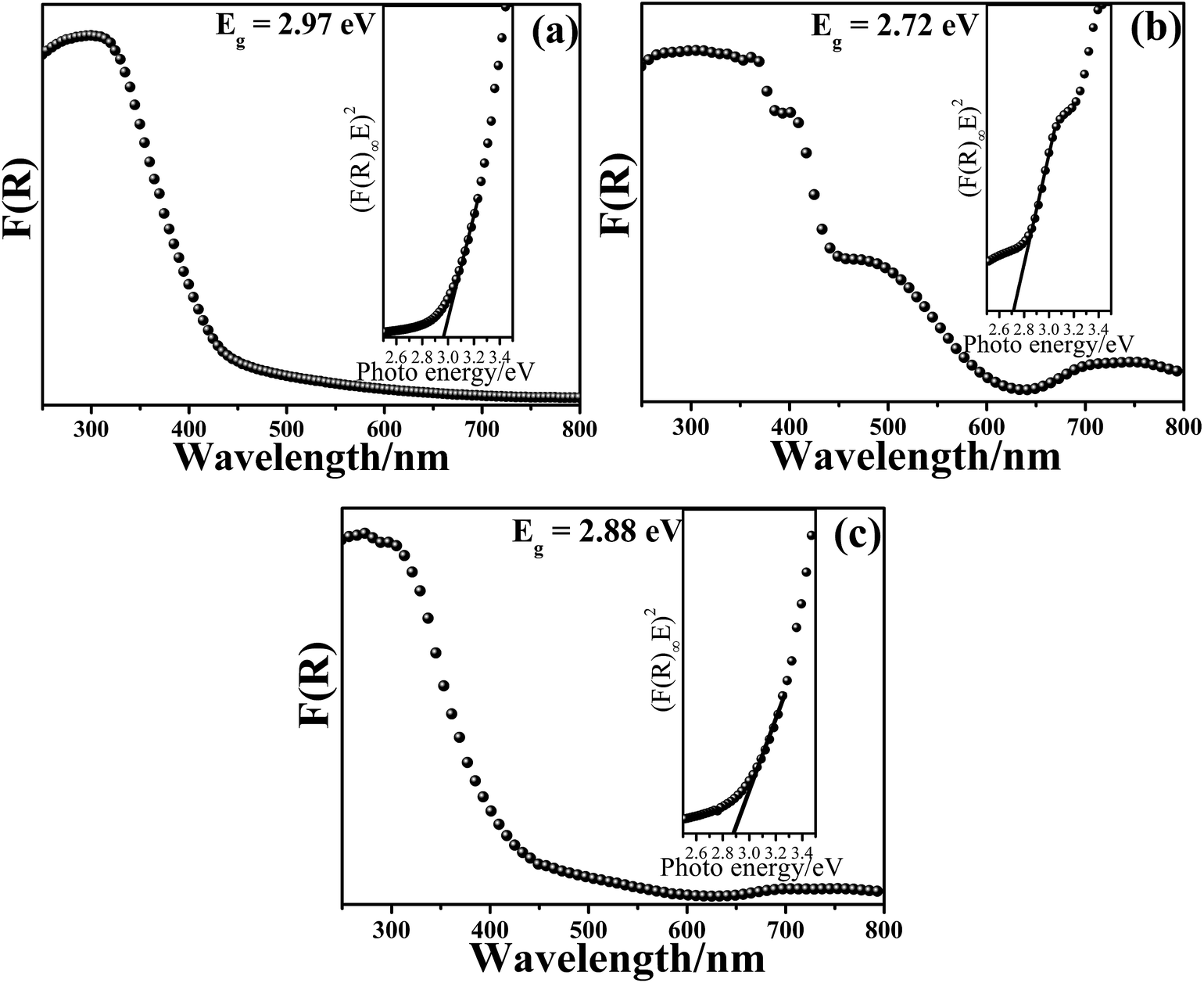 | ||
| Fig. 3 UV-vis diffuse reflectance spectrum and the plots of F(R) vs. photo energy for the estimation of the optical absorption edge energy of the products: (a) pure BWO, (b) pure BZO and (c) BZO–BWO heterostructures (S1). | ||
To demonstrate the potential application of the BZO–BWO heterostructures in the decomposition of organic pollutants, the photocatalytic activities of different photocatalysts were investigated by choosing the photocatalytic degradation of RhB with high concentrations as a model reaction (Fig. 4a). In our experiments, one can find that the concentration of RhB hardly changes after 90 min reaction in the absence of any photocatalyst. When bare BZO or BWO is employed as photocatalyst, the degradation rate of RhB is 10 and 23%, respectively. These show that the photocatalytic activity of single-component photocatalyst (BZO or BWO) is relatively low due to the high recombination rate of photo-generated carriers. When BZO and BWO are combined to construct BZO–BWO heterostructures, all three composite photocatalysts (S1, S2 and S3) exhibit excellent photocatalytic activity and S1 possesses the highest photocatalytic degradation efficiency (91%). The above experimental results indicate that the introduction of BWO into BZO can significantly inhibit the recombination of photo-induced electron–hole pairs and lead to the remarkable improvement of the photocatalytic activities. Recently, Shenawi-Khalil reported the preparation of BiOBr/bismuth oxyhydrate photocatalyst via a facile and simple hydrothermal method.35 Research indicated that excess bismuth oxyhydrate in the composite photocatalyst could act as a recombination center and impeded the improvement of photocatalytic activity, which might explain the difference of photocatalytic degradation efficiency in S1, S2 and S3. Furthermore, if the sample prepared by the physical mixture of BZO and BWO is used as photocatalyst, only 29% of RhB can be decomposed. Usually, composite photocatalyst obtained by physical mixing technology does not have intimate interfacial contacts between different components. So, it is difficult to realize the efficient separation of photo-generated charges as well as the remarkable improvement of photocatalytic efficiency, which proves the superiority of partial conversion strategy. To analyze in detail the photocatalysis kinetics of the RhB degradation, we apply the pseudo-first order model as expressed by eqn (3), which is usually used for photocatalytic degradation process if the initial concentration of pollutant is low.36–38
| ln(C0/C) = kt | (3) |
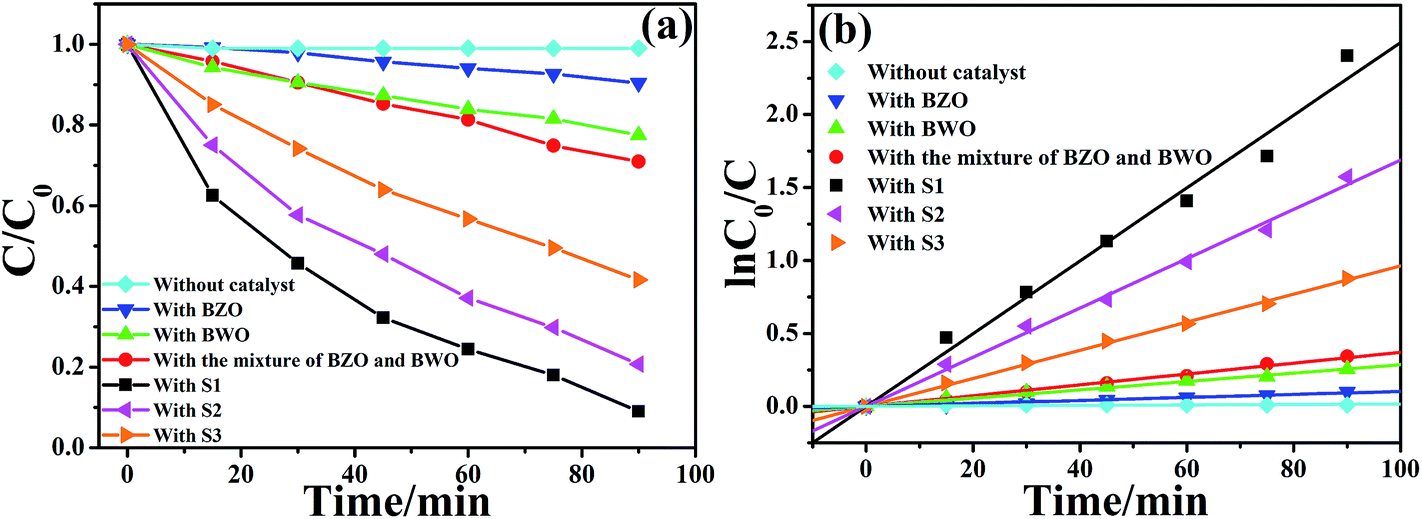 | ||
| Fig. 4 (a) Photodegradation efficiencies of RhB as a function of irradiation time and (b) kinetics of RhB decolorization in solutions. | ||
The C0 and C are the concentrations of organic dye in solution at time 0 and t, respectively, and k is the pseudo first-order rate constant. The rate constants calculated from the data plotted in Fig. 4a are summarized in Table 2. The experimental data obviously shows that the apparent rate constant k is 0.0010, 0.0029, 0.0037, 0.0250, 0.0169 and 0.0096 min−1 for pure BZO, BWO, physical mixture sample, S1, S2 and S3, respectively. In other words, BZO–BWO heterojunction photocatalysts (S1) exhibits the highest photocatalytic activity, which is almost 25.0, 8.6, 6.8, 1.5 and 2.6 times higher than those of pure BZO, BWO, physical mixture of BZO and BWO, S2 and S3, respectively.
| Series | Photocatalysts | First-order kinetic equation | k (min−1) | R |
|---|---|---|---|---|
| 1 | BZO | ln(C0/C) = 0.0010t | 0.0010 | 0.992 |
| 2 | BWO | ln(C0/C) = 0.0029t | 0.0029 | 0.996 |
| 3 | Mixture of BZO and BWO | ln(C0/C) = 0.0037t | 0.0037 | 0.997 |
| 4 | S1 | ln(C0/C) = 0.0250t | 0.0250 | 0.991 |
| 5 | S2 | ln(C0/C) = 0.0169t | 0.0169 | 0.997 |
| 6 | S3 | ln(C0/C) = 0.0096t | 0.0096 | 0.998 |
Photoluminescence (PL) spectrum arises from the migration, transfer, and separation efficiency of the photo-generated charge carriers in a semiconductor photocatalyst. In general, there is a close correlation between the photocatalytic activity and PL intensity. The lower the PL intensity, the lower the recombination rate of photo-induced carriers, and the higher the photocatalytic activity of photocatalysts.39–41 Fig. 5 shows the PL spectra of pure BZO, pure BWO, physical mixture of BZO and BWO, and BZO–BWO heterostructures (S1) at room temperature. It can be seen that BZO–BWO composite photocatalyst (S1) displays the lowest emission peak, suggesting that such heterojunctions can effectively inhibit the recombination of photogenerated charge carriers and possess the highest photocatalytic activity, which is in good agreement with the result from photodegradation experiment.
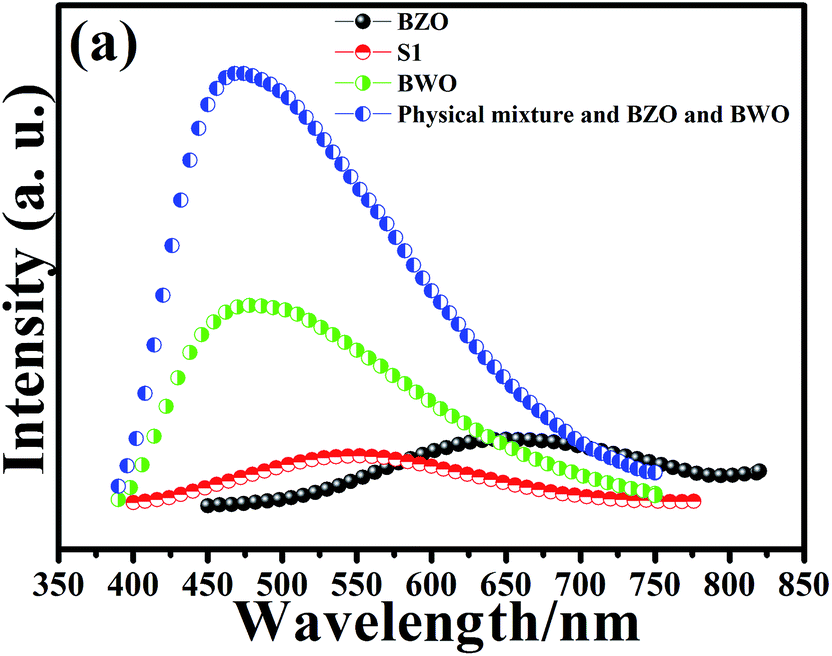 | ||
| Fig. 5 Room temperature PL spectra of different photocatalysts. | ||
To better understand the effect of the heterojunction on the high photocatalytic activity of the BZO–BWO heterostructure, we investigated its photoinduced charge transfer properties. Fig. 6 shows the photocurrent responses of BWO, BZO and BZO–BWO heterostructures (S1), under visible light irradiation. Experimental results reveal that BZO–BWO heterojunctions exhibit higher current density than the single-component semiconductors, which demonstrate that a more effective separation of photogenerated electron–hole pairs and faster interfacial charge transfer occur in the heterostructures.35
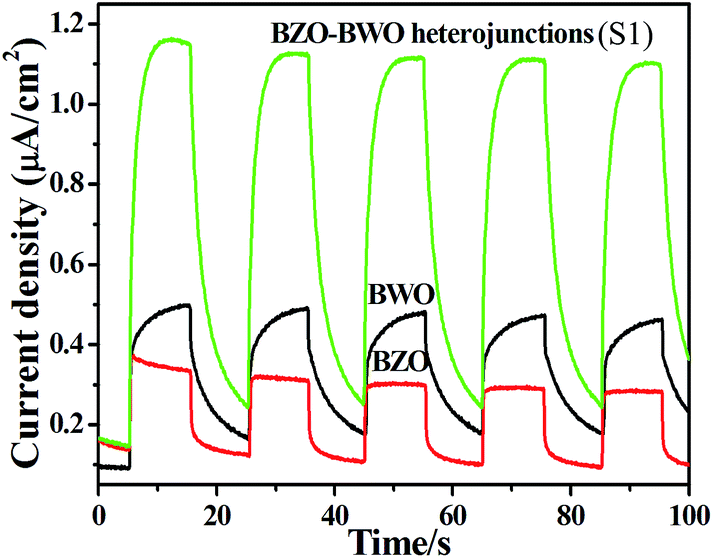 | ||
| Fig. 6 Photocurrent responses of different photocatalysts under visible-light irradiation. | ||
In order to study the photocatalytic degradation mechanism over the BZO–BWO composite photocatalyst (S1), some sacrificial agents such as isopropanol (IPA), sodium oxalate (Na2C2O4) and benzoquinone (BQ) were introduced (2.0 mM) into the RhB solution and served as scavengers of ˙OH, h+ and ˙O2−, respectively.42 The corresponding experimental results are shown in Fig. 7. Na2C2O4 has a strong influence on the degradation efficiency of RhB. However, the degradation rate of RhB slightly decreases after the addition of IPA and BQ. The above experimental results indicate that h+ played more important roles than ˙OH and ˙O2− in the photodegradation of RhB.
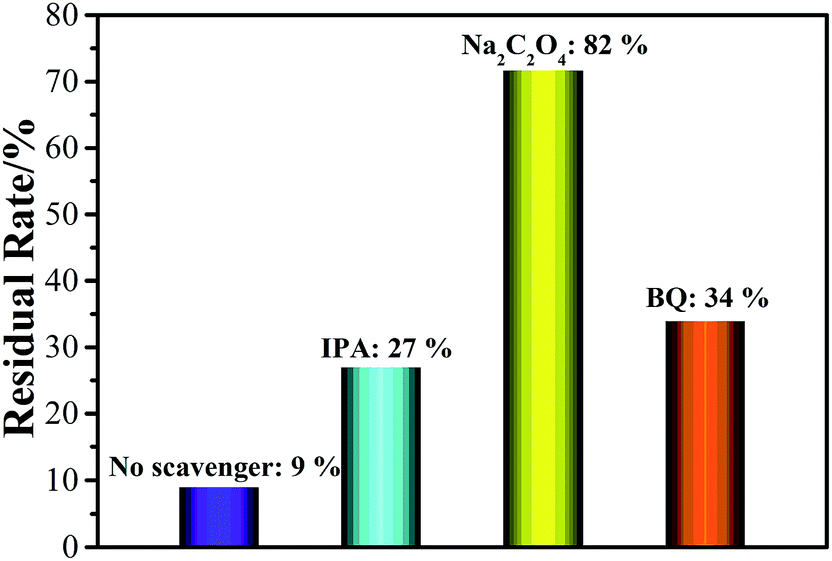 | ||
| Fig. 7 Photocatalytic degradation of RhB over the BZO–BWO heterostructures (S1) with or without quenchers. | ||
To fully understand the mechanism of the enhanced photocatalytic efficiency of the BZO–BWO heterostructures, the band edge positions of the valence and conduction band of the two semiconductors need to be determined. The conduction band bottoms are estimated in this study according to the below empirical formula:43
| ECB = X − 0.5Eg − E0 | (4) |
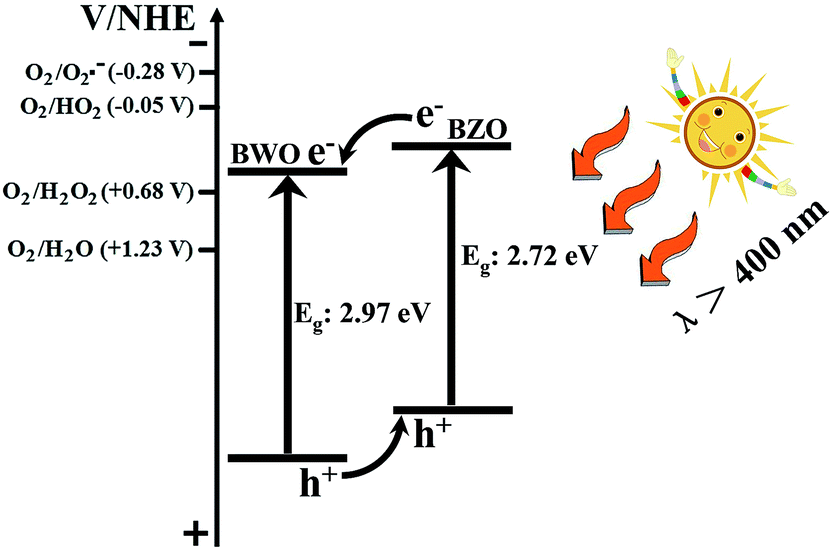 | ||
| Fig. 8 Diagram for energy band levels of BZO–BWO heterostructures and the possible charge separation process. | ||
4. Conclusions
In summary, visible-light-driven BZO–BWO heterostructure photocatalyst has been fabricated via a controllable partial conversion strategy. Photocatalytic experiments demonstrate that such heterojunctioned material exhibit higher photocatalytic activity than the single-component photocatalyst. The significant improvement of photocatalytic activity is closely associated with the efficient separation of photo-generated carriers resulting from the matching band positions and intimate interfacial contacts. This work reports a general and effective method to improve the visible light activity of sillenite-type photocatalytic materials and may find important applications in decomposition of organic wastewater.Acknowledgements
We gratefully acknowledge financial support from the Natural Foundation of Anhui Province (no. 1308085QB34 and 1408085QB31), the National Natural Science Foundation of China (no. 21201006 and 21301005) and Special Research Found for the Doctoral Program of Higher Education (no. 269 20123415120002).References
- Y. J. Wang, Q. S. Wang, X. Y. Zhan, F. M. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326 RSC.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455 CrossRef CAS PubMed.
- Y. Q. Qu and X. F. Duan, Chem. Soc. Rev., 2013, 42, 2568 RSC.
- J. G. Hou, S. Q. Jiao, H. M. Zhu and R. V. Kumar, CrystEngComm, 2011, 13, 4735 RSC.
- C. Zhou, Y. F. Zhao, T. Bian, L. Shang, H. J. Yu, L. Z. Wu, C. H. Tung and T. R. Zhang, Chem. Commun., 2013, 49, 9872 RSC.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217 CAS.
- W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946 RSC.
- H. Tong, S. X. Ouyang, Y. P. Bi, N. Umezawa, M. Oshikiri and J. H. Ye, Adv. Mater., 2012, 24, 229 CrossRef CAS PubMed.
- Y. G. Wang, R. He, M. Yang, T. Wen, H. Zhang, J. Liang, Z. S. Lin, Y. X. Wang, G. B. Li and J. H. Lin, CrystEngComm, 2012, 14, 1063 RSC.
- L. J. Xie, J. F. Ma, H. Tian, J. Zhou, Z. Q. Zhao, P. W. Wu, Y. M. Hu, Y. G. Wang, J. T. Tao and X. Y. Zhu, Mater. Lett., 2006, 60, 284 CrossRef CAS PubMed.
- W. Wei, Y. Dai and B. B. Huang, J. Phys. Chem. C, 2009, 113, 5658 CAS.
- J. B. Lu, Y. Dai, Y. T. Zhu and B. B. Huang, ChemCatChem, 2011, 3, 378 CrossRef CAS.
- W. Guo, S. Q. Zhang, Y. N. Guo, L. Ma, F. Su, Y. H. Guo and A. F. Geng, RSC Adv., 2013, 3, 4008 RSC.
- D. F. Hou, X. L. Hu, Y. W. Wen, B. Shan, P. Hu, X. Q. Xiong, Y. Qiao and Y. H. Huang, Phys. Chem. Chem. Phys., 2013, 15, 20698 RSC.
- A. M. L. Lopes, J. P. Araújo and S. Ferdov, Dalton Trans., 2014, 43, 18010 RSC.
- J. G. Hou, Y. F. Qu, D. Krsmanovic, C. Ducati, D. Eder and R. V. Kumar, Chem. Commun., 2009, 3937 RSC.
- J. G. Hou, Y. F. Qu, D. Krsmanovic, C. Ducati, D. Eder and R. V. Kumar, J. Mater. Chem., 2010, 20, 2418 RSC.
- Z. Wan and G. K. Zhang, Sci. Rep., 2014, 4, 6298 CrossRef CAS PubMed.
- W. F. Yao, H. Wang, X. H. Xu, J. T. Zhou, X. N. Yang, Y. Zhang, S. X. Shang and M. Wang, Chem. Phys. Lett., 2003, 377, 501 CrossRef CAS.
- W. F. Yao, X. H. Xu, J. T. Zhou, X. N. Yang, Y. Zhang, S. X. Shang, H. Wang and B. B. Huang, J. Mol. Catal. A: Chem., 2004, 212, 323 CrossRef CAS PubMed.
- B. Lal, S. K. Patro and S. Singh, J. Sol-Gel Sci. Technol., 2010, 56, 340 CrossRef CAS.
- R. Wisedsri, T. Chaisuwan and S. Wongkasemjit, Mater. Lett., 2011, 65, 3237 CrossRef CAS PubMed.
- J. W. Tang and J. H. Ye, Chem. Phys. Lett., 2005, 410, 104 CrossRef CAS PubMed.
- J. G. Hou, R. Cao, S. Q. Jiao, H. M. Zhu and R. V. Kumar, Appl. Catal., B, 2011, 104, 399 CrossRef CAS PubMed.
- W. Guo, Y. X. Yang, Y. N. Guo, Y. Q. Jia, H. B. Liu and Y. H. Guo, Phys. Chem. Chem. Phys., 2014, 16, 2705 RSC.
- X. N. Li, R. K. Huang, Y. H. Hu, Y. J. Chen, W. J. Liu, R. S. Yuan and Z. H. Li, Inorg. Chem., 2012, 51, 6245 CrossRef CAS PubMed.
- A. W. Sun, H. Chen, C. Y. Song, F. Jiang, X. Wang and Y. S. Fu, RSC Adv., 2013, 3, 4332 RSC.
- L. H. Yu, Y. Huang, G. C. Xiao and D. Z. Li, J. Mater. Chem. A, 2013, 1, 9637 CAS.
- J. G. Hou, Z. Wang, S. Q. Jiao and H. M. Zhu, J. Hazard. Mater., 2011, 192, 1772 CrossRef CAS PubMed.
- H. F. Cheng, B. B. Huang, X. Y. Qin, X. Y. Zhang and Y. Dai, Chem. Commun., 2012, 48, 97 RSC.
- H. F. Cheng, B. B. Huang, Y. Y. Liu, Z. Y. Wang, X. Y. Qin, X. Y. Zhang and Y. Dai, Chem. Commun., 2012, 48, 9729 RSC.
- F. Dong, X. Feng, Y. X. Zhang, C. F. Gao and Z. B. Wu, RSC Adv., 2015, 5, 11714–11723 RSC.
- J. Wu, F. Duan, Y. Zheng and Y. Xie, J. Phys. Chem. C, 2007, 111, 12866 CAS.
- S. Shenawi-Khalil, V. Uvarov, S. Fronton, I. Popov and Y. Sasson, J. Phys. Chem. C, 2012, 116, 11004 CAS.
- L. Zhang, X. F. Cao, X. T. Chen and Z. L. Xue, J. Colloid Interface Sci., 2011, 354, 630 CrossRef CAS PubMed.
- Z. Wan, G. K. Zhang, J. T. Wang and Y. L. Zhang, RSC Adv., 2013, 3, 19617 RSC.
- L. Y. Huang, H. Xu, Y. P. Li, H. M. Li, X. N. Cheng, J. X. Xia, Y. G. Xu and G. B. Cai, Dalton Trans., 2013, 42, 8606 RSC.
- Z. J. Zhang, W. Z. Wang, L. Wang and S. M. Sun, ACS Appl. Mater. Interfaces, 2012, 4, 593 CAS.
- G. H. Tian, Y. J. Chen, J. Zhou, C. G. Tian, R. Li, C. J. Wang and H. G. Fu, CrystEngComm, 2014, 16, 842 RSC.
- D. L. Jiang, L. L. Chen, J. J. Zhu, M. Chen, W. D. Shi and J. M. Xie, Dalton Trans., 2013, 42, 15726 RSC.
- Y. Xu, S. C. Xu, S. Wang, Y. X. Zhang and G. H. Li, Dalton Trans., 2014, 43, 479 RSC.
- R. Bajaj, M. Sharma and D. Bahadur, Dalton Trans., 2013, 42, 6736 RSC.
- J. Jiang, X. Zhang, P. B. Sun and L. Z. Zhang, J. Phys. Chem. C, 2011, 115, 20555 CAS.
- H. W. Huang, S. B. Wang, N. Tian and Y. H. Zhang, RSC Adv., 2014, 4, 5561 RSC.
- M. L. Guan, D. K. Ma, S. W. Hu, Y. J. Chen and S. M. Huang, Inorg. Chem., 2011, 50, 800 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01327e |
| This journal is © The Royal Society of Chemistry 2015 |