
Insight into the structural and functional features of myoglobin from Hystrix cristata L. and Rangifer tarandus L.†
Abstract
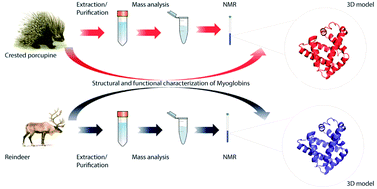
The amino acid sequence, structural and functional features of two novel myoglobins (Mbs) isolated from a crested porcupine (Hystrix cristata L.) and reindeer (Rangifer tarandus L.) were determined. The primary structure was achieved by using a combined approach based on de novo sequencing by ESI-Q-TOF MS/MS and peptide mapping by MALDI-TOF MS. This strategy allowed us to determine the primary structure of crested porcupine and reindeer Mbs. To go deeper, 3D modeling studies followed by structural characterization by NMR on both myoglobins demonstrate that reindeer Mb shows slightly different orientation of F, G and H α-helices. As a consequence, reindeer Mb may differently modulate the heme environment, facilitating oxygenation as well as ensuring that the heme iron remains in a ferrous state. Finally, reindeer Mb shows a less stable conformation with respect to crested porcupine Mb (Tm 353.7 K vs. Tm 356.3 K, respectively).
 Please wait while we load your content...
Please wait while we load your content...

