
Electrochemical exfoliation of graphite to produce graphene using tetrasodium pyrophosphate
Abstract
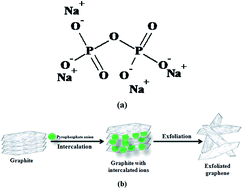
An electrochemical exfoliation based synthetic methodology to produce graphene is provided. An eco-friendly and non-toxic tetrasodium pyrophosphate solution in which the pyrophosphate anion acts as an intercalating ion was used as the electroactive media. Five different ion intercalation potentials were used. Characterization by microscopy, X-ray diffraction, Raman spectroscopy and UV-Visible spectroscopic techniques confirmed that all the potentials produced nano to micrometer sized graphene sheets. No trace of graphene oxide was detected. It was observed that (i) an increase in the intercalation potential increased the graphene yield and (ii) the defect density of graphene did not change significantly with a change in the intercalation potential.
 Please wait while we load your content...
Please wait while we load your content...

