
Formation of PCP pincer cobalt complexes with cobaltacyclopropane moieties via double Csp3–H bond activation†
Abstract
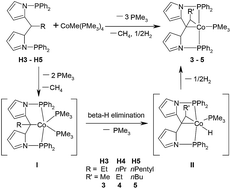
The introduction and changes of the substituents at the middle carbon atom of the preligand dipyrrolmethane have a significant impact on the reaction results. When the substituent at the Csp3 atom is a methyl group, the reaction of the preligand with CoMe(PMe3)4 delivered cobalt(I) complex 2 as a Csp3–H bond activation product. In the case of ethyl, propyl and pentyl groups, PCP pincer cobalt complexes 3–5 with cobaltacyclopropane moieties were formed via double Csp3–H bond activation. With iso-propyl as the substituent, cobalt(I) complex 6 as Csp2–H activation product was obtained.
 Please wait while we load your content...
Please wait while we load your content...

