
Plasmodium falciparum subtilisin-like protease 1: discovery of potent difluorostatone-based inhibitors†
Abstract
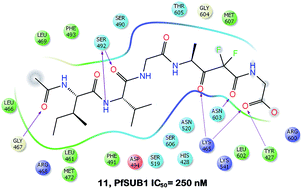
Currently available drugs to treat malaria are often ineffective due to the acquisition of drug resistance. In this context, drugs with innovative modes of action and no liability to cross-resistance are urgently needed. Recently, subtilisin-like protease 1, a P. falciparum serine protease involved in merozoite egress from red blood cells and invasion, has been identified as potential drug target. We describe herein the development of a series of potent PfSUB1 inhibitors. Combining a straightforward synthetic approach, an in depth structure–activity study and in silico investigation, we identified the most potent inhibitors known to date, characterized by an improved enzyme inhibitory potency and a reduced peptidic character over the prototypic peptides.
 Please wait while we load your content...
Please wait while we load your content...

