
Energy decomposition analysis of gauche preference in 2-haloethanol, 2-haloethylamine (halogen = F, Cl), their protonated forms and anti preference in 1-chloro-2-fluoroethane†
Marija Baranac-Stojanović*a,
Jovana Aleksićb and
Milovan Stojanovićb
aFaculty of Chemistry, University of Belgrade, Studentski trg 12-16, P. O. Box 158, 11000 Belgrade, Serbia. E-mail: mbaranac@chem.bg.ac.rs
bCenter for Chemistry ICTM, University of Belgrade, P. O. Box 473, 11000 Belgrade, Serbia
First published on 25th February 2015
Abstract
2-Haloethanol and 2-haloethylamine (halogen = F, Cl) prefer gauche conformation. This preference is significantly increased upon protonation. Commonly used explanations are based on intramolecular hydrogen bonding and hyperconjugation. 1,2-Difluoroethane prefers gauche conformation, too, while gaseous 1-chloro-2-fluoroethane is more stable as the anti conformer. The origin of these conformational preferences has been investigated by a quantitative partitioning of the gauche/anti energy difference into contributions from electrostatic, orbital, dispersion and Pauli interactions, and structural changes accompanying the rotation. The results show that, with two exceptions, the most important contributor to the gauche preference is electrostatic attraction, which is larger in gauche forms relative to the anti ones. Next come orbital interactions, while dispersion forces provide the smallest stabilizing energy. These energy components override destabilizing Pauli interactions and energetically costly structural changes. All gauche preferences also benefit from stereoelectronic effects, except in protonated 2-chloroethanol which, instead, shows a significant Cl lone pair → O–H antibond mixing, associated with hydrogen bonding. The increase in the Pauli repulsion upon anti to gauche isomerization is more pronounced for fluorine than for chlorine derivatives. Thus, the smaller gauche effect in chloro-compounds and the anti preference in 1-chloro-2-fluoroethane have their origin in the decrease in electrostatic and orbital stabilizing interactions, a drop in the former being more pronounced.
Introduction
Fluorine is the most electronegative atom in the periodic table and thus the C–F bond is the most polar bond in organic chemistry.1 Therefore, the observation that 1,2-difluoroethane (DFE) prefers gauche conformation over the anti by 0.5–1 kcal mol−1 (ref. 2 and 3) is surprising, taking into account the expected strong repulsive interaction between the two C–F bond dipoles in the gauche form. This gauche preference, also termed as gauche effect,4 has been rationalized by the bent bond model of Wiberg et al.5 according to which electronegative fluorine atoms cause C–C bond bending in both anti and gauche forms, but leading to poorer orbital overlap in the former. Another, more widely used explanation is based on hyperconjugation. Thus, the two vicinal σC–H → σ*C–F hyperconjugative interactions are involved in the stabilization of the gauche conformer.3c,d,6 Recently, we proposed that gauche-DFE also benefits from more favourable electrostatic interactions, that is, this type of interactions should be considered as an all-charge phenomenon rather than partial interaction between pairs of bonds.7 This finding was in accord with an earlier explanation of the gauche effect in various compounds as a prevailing nucleus–electron attraction over nucleus–nucleus and electron–electron repulsion.4The fluorine gauche effect was found experimentally and/or theoretically in various other compounds having an electronegative substituent in β-position to the fluorine atom, such as O-β-fluoroethyl esters,8 N-β-fluoroethylamides,9 1-fluoro-2-nitroethane,10 1-azido-2-fluoroethane,10 1-fluoro-2-iso(thio)cyanatoethane,10 2-fluoroimines,11 2-fluoroethanol12 and 2-fluoroethylamine.12e,13 Its origin is commonly ascribed to stereoelectronic effects, that is, the stabilizing hyperconjugative interactions wherein σ*C–F and σ*C–X orbitals (X = electronegative atom in β-position to F) act as acceptors of electron density donated from antiperiplanar σC–H bonds in gauche forms. Electrostatic attraction between fluorine and an electropositive atom contained in a β-substituent may add additional stabilization.10 Although, the conformational behaviour of 2-fluoroethanol (FE) has been ascribed to intramolecular hydrogen bonding interactions12a rather than stereoelectronic effects,12b,e this explanation was questioned12h based on the facts that organic fluorine is poor hydrogen bond acceptor.14 In particular, fluorine atom hardly accepts intramolecular five-membered F⋯H–O hydrogen bond due to geometric restrictions, which do not allow sufficiently close contact between F and H atoms.12h,15 Instead, the gauche prevalence in FE may stem from an electrostatic attraction between antiparallel OH and CF bond dipoles12d and stronger hyperconjugative interactions.12f Although, a repulsion between the lone pair electrons on oxygen and fluorine atoms could also turn the hydroxyl hydrogen atom toward the fluorine.12c The larger stability of gauche forms in 2-fluoroethylamine (FEA) has also been attributed to the formation of intramolecular hydrogen bonds,12e,13b seen as an electrostatic attractive interaction between the N–H and C–F bond dipoles, rather than covalent bonding.13a
Protonation of FEA increases gauche preference from 1.0 kcal mol−1 in neutral amine to 5.8 kcal mol−1 in 2-fluoroethylammonium ion (FEAH), while protonation of FE results in gauche preference of 7.2 kcal mol−1 relative to 2.0 kcal mol−1 for the alcohol.12e Both stereoelectronic effects and intramolecular hydrogen bonding interactions were invoked to explain the calculated large stability of gauche conformers relative to their anti counterparts.12e However, X-ray structure analysis of 2-fluoroethylammonium chloride revealed no intramolecular F⋯H–N hydrogen bonding interaction.12e Later theoretical work showed that the strength of hyperconjugative interactions is similar in gauche-FEA and its protonated structure, and the larger stabilization of gauche-FEAH vs. anti-FEAH as compared to that of gauche-FEA vs. anti-FEA was attributed to strong intramolecular F⋯H–N hydrogen bonding in the protonated gauche form.13c An additional explanation is based on electrostatic interactions.10 The fluorine gauche effect extends to the related (a)cyclic systems containing positively charged nitrogen atom and is ascribed to the strong through space charge–dipole or dipole–dipole (+N–H and C–F) attractive interaction, the strength of which compares with that of a reasonably strong hydrogen bond.16
Hence, introduction of fluorine atom β to an electronegative substituent has important consequences on conformational behaviour and has emerged as a powerful tool in synthetic organic chemistry.17
The replacement of fluorine with chlorine atom in DFE results in a loss of gauche preference in 1-chloro-2-fluoroethane (CFE) in the gas phase.3a,12d,18 The same replacement in FE and FEA retains the gauche preference in 2-chloroethanol (CE)12d,f,g,19 and 2-chloroethylamine (CEA),20 but decreases the energy differences between gauche and anti forms relative to those for fluorine compounds. In both CE and CEA, the dominance of gauche conformers has been attributed to stabilizing intramolecular hydrogen bonding interactions19a usually described as an electrostatic attraction between antiparallel C–Cl and O(N)–H bond dipoles.12d,f,19b,c,20 The operation of hyperconjugative interactions was invoked, too.12f The enhanced stability of the anti-CFE relative to the gauche-CFE was rationalized as a combination of electrostatic repulsion between the C–F and C–Cl bond dipoles and a weaker hyperconjugative preference for gauche form.3a A smaller tendency of chlorinated compounds for gauche conformation compared with fluoro derivatives could be anticipated on the basis of chlorine's larger size (van der Waals radii of F and Cl atoms are 1.47 Å and 1.75 Å, respectively),21 thus introducing larger steric repulsion in gauche forms. The electrostatic repulsion between bond dipoles in gauche forms is expected to be smaller. Though, other factors, such as hyperconjugation, should play a role, as well. Unlike fluoro derivatives, chlorine-containing molecules have been less studied with respect to the origin of their conformational preferences.
Understanding the factors governing conformational behaviour is crucial to address various questions in organic chemistry and biochemistry, since molecular properties, reactivity and interactions with other molecules are influenced by their conformation. The purpose of this study is to quantitatively decompose the energy difference between gauche and anti conformers of FE, CE, FEA, CEA and their protonated forms into contributions from electrostatic, orbital and dispersion interactions, Pauli repulsion and energy required for structural changes and thus to get a further insight into the origin of gauche effect in these molecules. One aim is also to explain the weaker tendency for gauche conformations in the chloro derivatives. For the latter to be done, CFE and DFE were also included in the study. The results would deepen our knowledge about the origin of conformational behaviour of the mentioned and related molecules, the importance of which is also reflected in the increasing application of fluorine compounds in organic and medicinal chemistry.11,16b,17 To the best of our knowledge, no such an attempt has been made, though quantifications of some of the interactions considered to be involved have been done. These include hyperconjugative interactions and intramolecular hydrogen bonding/antiparallel dipoles attraction, often estimated on the basis of the natural bond orbital (NBO) analysis10,12d,f,h,13c or from the relative energies of corresponding conformers.12b,e,19a
Computational details
Various conformers of studied compounds were fully optimized at the MP2/6-311++G** level22 using the Gaussian 09 program package.23 They were characterized as energy minima by the absence of imaginary frequencies. In the following conformational energy analysis, done at the same level of theory, overall molecules have been built from the two radicals XH2C˙ (X = F, Cl) and ˙CH2Y (Y = F, OH, +OH2, NH2, +NH3), taken with opposite spins (α and β superscripts in Scheme 1) so that they can form a bond. | ||
| Scheme 1 | ||
Total binding energy ΔE between them consists of two major parts (eqn (1)):
| ΔE = ΔEprep + ΔEint | (1) |
In the equation, the preparation energy ΔEprep represents the amount of energy required to deform two isolated radicals (XH2C˙ and ˙CH2Y) from their equilibrium geometry to the geometry they adopt in the final molecules. The interaction energy ΔEint is the energy change occurring when prepared (deformed) fragments XH2C˙ and ˙CH2Y combine to form the molecule. The latter energy term can be further decomposed into five components (eqn (2)) by using the localized molecular orbital energy decomposition analysis (LMOEDA),24 implemented in the Gamess program package.25
| ΔEint = ΔEelstat + ΔEex + ΔErep + ΔEpol + ΔEdisp | (2) |
The electrostatic energy ΔEelstat corresponds to nucleus–nucleus and electron–electron repulsion, and nucleus–electron attraction between the two prepared radical fragments that adopt their positions in the final molecule, and is usually stabilizing (attractive). The exchange energy ΔEex refers to the quantum mechanical exchange between the same-spin electrons and is simultaneously counteracted by the repulsion energy ΔErep. Taken together, they form the exchange repulsion26 or Pauli repulsion27 of other EDA schemes, which is a destabilizing interaction, also referred to, herein, as the steric repulsion. This type of repulsion is caused by the fact that two electrons with the same spin cannot occupy the same region in space. The polarization energy ΔEpol is an orbital relaxation energy which comes from a change in orbital shapes upon binding and also includes empty-occupied orbital mixing within one fragment due to the presence of another (polarization) and between two fragments (charge transfer). The dispersion energy term ΔEdisp at the MP2 level comes from electron correlation correction to the Hartree–Fock interaction energy. The energy change that follows anti → gauche rotation is calculated as a difference between total binding energies of gauche and anti conformers, where the change in the ΔEprep reflects gain or loss in energy due to the structural changes that accompany the conformational isomerization, whereas the change in the ΔEint is associated with the change in the nature of chemical bonding. Such an analysis of the interaction energy between two or more fragments constituting a molecule has been applied before to study the rotational barrier in ethane24,28 and in group 13-elements (E = B – Tl),29 distortion to the trans-bent geometry in heavier ethylene homologues,30 the isomerization energy of heterocyclic31 and polycyclic32 compounds, the strength of conjugation and hyperconjugation,33 and the nature of covalent bonds.34
Energies of hyperconjugative interactions were calculated by using the natural bond orbital (NBO) analysis (NBO version 6.0 (ref. 35) linked to G09 was employed).
Results and discussion
1,2-Difluoroethane and 1-chloro-2-fluoroethane
The MP2/6-311++G** optimized bond lengths, bond angles and torsional angles for anti and gauche conformers of 1,2-difluoroethane (DFE) and 1-chloro-2-fluoroethane (CFE) are given in Tables S1 and S2 in the ESI.† They are very close to the experimental values obtained from infrared36 and microwave37 spectroscopy, and electron diffraction.18a In the case of DFE, gauche conformer is more stable than anti by ΔE = −0.77 kcal mol−1 (a-DFE → g-DFE in Fig. 1 and Table 1). This is in accord with previous experimental and theoretical results.2,3 By contrast, CFE prefers anti conformation by ΔE = 0.52 kcal mol−1 (a-CFE → g-CFE in Fig. 1 and Table 1), which also compares with previous estimates in the gas phase.3a,12d,18 Table 1 lists the energy decomposition results for anti and gauche conformers of DFE and CFE (a-DFE, g-DFE, a-CFE and g-CFE) and for the anti → gauche rotation of both molecules (a-DFE → g-DFE and a-CFE → g-CFE). | ||
| Fig. 1 Energy minimum conformations of 1,2-difluoroethane (DFE) and 2-chloro-1-fluoroethane (CFE), their relative electronic energies (kcal mol−1) and energy changes occurring upon anti → gauche rotation. | ||
| Conformation | ΔEelstat | ΔEex+rep | ΔEpol | ΔEdisp | ΔEint | ΔEprep | ΔE |
|---|---|---|---|---|---|---|---|
| a ΔEelstat = electrostatic energy, ΔEex+rep = exchange repulsion energy, ΔEpol = polarization energy, ΔEdisp = dispersion energy, ΔEint = interaction energy, ΔEprep = preparation energy, ΔE = total binding energy. | |||||||
| a-DFE | −148.82 | 221.67 | −155.48 | −25.50 | −108.13 | 10.31 | −97.82 |
| g-DFE | −152.20 | 229.24 | −160.01 | −26.11 | −109.08 | 10.50 | −98.58 |
| a-DFE → g-DFE | −3.38 | 7.57 | −4.53 | −0.61 | −0.95 | 0.18 | −0.77 |
| a-CFE | −149.20 | 225.17 | −156.91 | −29.12 | −110.06 | 12.05 | −98.01 |
| g-CFE | −150.02 | 228.83 | −159.04 | −29.49 | −109.72 | 12.24 | −97.48 |
| a-CFE → g-CFE | −0.82 | 3.66 | −2.13 | −0.37 | 0.34 | 0.18 | 0.52 |
Our MP2 results for DFE agree with previously reported B3LYP/6-311+G** results7 that the conformational preference in DFE comes from the more favourable interaction energy term ΔEint = −0.95 kcal mol−1, while the preparation energy slightly disfavours the gauche conformation. Somewhat unexpectedly, data in Table 1, like those in the previous publication,7 show that g-DFE is stabilized not only by more favourable orbital interactions (ΔEpol = −4.53 kcal mol−1), but also by more attractive electrostatic interactions (ΔEelstat = −3.38 kcal mol−1). This finding points to the conclusion that electrostatic interactions should be considered as an all-charge phenomenon, rather than partial interactions between pairs of bonds. Dispersion energy ΔEdisp = −0.61 kcal mol−1, too, slightly favours gauche conformation and the only destabilizing interaction in the ΔEint energy part is steric repulsion coming from Pauli interactions.
In CFE, gauche conformer is disfavoured by both ΔEint = 0.34 kcal mol−1 and ΔEprep = 0.18 kcal mol−1. It is of interest to analyze the origin of destabilizing ΔEint compared to the stabilizing ΔEint in DFE. An intuitive explanation for the reversal of conformational preferences in DFE and CFE would be a stronger steric repulsion between the more voluminous chlorine and the gauche oriented fluorine atom in g-CFE than between the two fluorine atoms in g-DFE. As Table 1 shows, the steric repulsion indeed increases when a-CFE rotates to g-CFE (ΔEex+rep = 3.66 kcal mol−1), but this energy rise is smaller than that for DFE (ΔEex+rep = 7.57 kcal mol−1). It should be noted that this steric repulsion reflects the all-electron repulsive interactions, not just those between halogen atoms which are by 0.06 Å closer to each other in DFE than their van der Waals radii would allow, and by 0.10 Å in CFE. Thus, the steric repulsion cannot account for the reversal of conformational preference in CFE compared to that in DFE. The data in Table 1 show that the nature of stabilizing interactions is practically the same in two conformers of both DFE and CFE: ΔEpol is the largest attractive component contributing about 47% of all attractive forces, next comes ΔEelstat (about 45%), while ΔEdisp contributes the smallest stabilization (about 8%). However, their magnitude differ. Upon a → g rotation, these three interactions provide much more stabilization in the case of DFE, which overcomes the destabilizing steric repulsion and leads to the overall negative interaction energy (stabilization of gauche conformer). In the case of CFE, the change in the magnitude of stabilizing interactions upon a → g isomerization is not large enough to overcome the increase in the steric repulsion. Thus, it is a drop in electrostatic, orbital and dispersion interactions, following the isomerization, which is responsible for the anti preference in isolated CFE. The energy gain due to structural changes occurring upon isomerization is the same in both molecules (ΔEprep in Table 1).
Since the LMOEDA does not allow separation of charge transfer interactions from other orbital interactions, vicinal hyperconjugative interactions, often invoked to explain gauche preferences,3c,d,6,10 were estimated by using the NBO analysis, at the HF/6-311++G** level. Within the NBO framework, the stabilizing energy gained from delocalization of electron density from filled orbitals to empty antibonding orbitals can be calculated by a second-order perturbation approach (referred to as E(2) energies) or by deletion of the corresponding off-diagonal elements of the Fock matrix in the NBO basis and recalculating the energy (referred to as E(del) energies).38 We used both approaches for the calculation of all vicinal hyperconjugative interactions (synclinal and anti) between XCH2 and CH2Y fragments. For E(2) energies, this corresponds to the sum of all vicinal interactions. For E(del) energies, we set to zero all Fock matrix elements corresponding to delocalizing interactions from C–H and C–X orbitals on one fragment into the antibonding C–H and C–Y orbitals on another fragment, and vice versa (deletion type 3 in NBO Manual, p. B-17).38c The calculated values correlate well with each other for all molecules considered, as has also been shown for other systems.6b,38,39 Data for DFE and CFE are given in Table 2 as E(2)synclinal/anti and E(del)synclinal/anti, respectively, and for other compounds in tables that will be discussed later. A change in hyperconjugative interaction energy occurring upon anti → gauche isomerization is denoted as E(2)synclinal/anti a → g and E(del)synclinal/anti a → g in all tables considering these interactions. The more positive value means stronger interaction.
| a-DFE | g-DFE | a-CFE | g-CFE | |
|---|---|---|---|---|
| Vicinal hyperconjugation | ||||
| E(2)synclinal/anti | 18.48 | 23.96 | 21.32 | 25.11 |
| E(2)synclinal/anti a → g | 5.48 | 3.79 | ||
| E(del)synclinal/anti | 17.38 | 21.98 | 20.17 | 22.96 |
| E(del)synclinal/anti a → g | 4.60 | 2.78 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Anti hyperconjugation | ||||
| C–H → C–H* | 2.59 (×4) | 2.81 (×2) | 3.08 (×2) | 3.15 |
| 2.73 (×2) | 2.95 | |||
| C–F → C–Y* | 1.80 | 2.22 | ||
| C–Y → C–F* | 1.80 | 3.76 | ||
| C–H → C–F* | 5.81 | 5.46 | ||
| C–H → C–Y* | 5.81 | 6.82 | ||
| C–F → C–H* | 0.85 | 1.06 | ||
| C–Y → C–H* | 0.85 | 1.91 | ||
| Total E(2)anti | 13.96 | 18.94 | 17.60 | 21.35 |
| Total E(2)anti a → g | 4.98 | 3.75 | ||
The results from the second-order perturbation approach (individual E(2) values) showed that anti hyperconjugative interactions account for the majority of these charge transfer interactions and they are the most affected by rotation. The synclinal interactions change little upon rotation for all studied molecules (less than 0.5 kcal mol−1), whereas changes in anti interactions can reach values of 6.4 kcal mol−1. For these reasons, only individual anti hyperconjugative interactions obtained from the second-order perturbation approach are shown in tables, where total E(2)anti values represent the sum of the six interactions for corresponding conformers, and total E(2)anti a → g values show their change upon the rotation.
The results in Table 2 show that gauche conformers of both DFE and CFE are more stabilized by hyperconjugation than their anti counterparts, so that stereoelectronic effects favour gauche forms. The most important contributions come from C–H → C–F* and C–H → C–Cl* delocalizations. The latter one is more stabilizing, which is in accord with the stronger electron-accepting ability of the C–Cl* antibond vs. C–F* antibond, due to its lower energy.6b There is an increase in the hyperconjugative stabilization in g-CFE relative to g-DFE. This increase originates mainly from the larger C–H → C–Cl* and C–Cl → C–H* delocalizations (E(2)C–H → C–Cl* = 6.82 kcal mol−1 vs. E(2)C–H → C–F* = 5.81 kcal mol−1, E(2)C–Cl → C–H* = 1.91 kcal mol−1 vs. E(2)C–F → C–H* = 0.85 kcal mol−1), showing that the C–Cl bond is also a better electron-donor due to its higher energy relative to energy of the C–F bond orbital. The anti conformer of CFE is also better stabilized by hyperconjugation than is anti conformer of DFE, mostly because of an increased C–Cl → C–F* interaction (3.76 kcal mol−1) in a-CFE compared to the C–F → C–F* one in a-DFE (1.80 kcal mol−1), though other interactions contribute, as well. The increase in hyperconjugative stabilization in a-CFE vs. a-DFE exceeds the increase in hyperconjugative stabilization in g-CFE vs. g-DFE. As a result, the anti → gauche conformational change benefits less from hyperconjugation, both total and anti, in CFE compared to DFE (Table 2). Overall, both hyperconjugative and all orbital interactions (ΔEpol in Table 1) contribute less stabilizing energy to the anti → gauche conformational isomerization in CFE with respect to that in DFE.
Thus, it is a drop in electrostatic and orbital stabilizing interactions which is responsible for the anti preference of isolated CFE. Despite the expected electrostatic repulsion between the two C–F bond dipoles in DFE and the C–F and C–Cl bond dipoles in CFE in their gauche conformers, they are actually more stabilized by electrostatic interactions than the anti forms. Somewhat counterintuitively, this stabilization is larger in DFE (ΔEelstat = −3.38 kcal mol−1, Table 1) than in CFE (ΔEelstat = −0.82 kcal mol−1, Table 1). These findings lead to conclusion that small electronegative atom, such as fluorine, contributes more electrostatic stabilization to the anti → gauche isomerization than does the less electronegative and bulkier atom, such as chlorine. The above conclusion also supports an earlier explanation of the gauche effect in terms of electrostatic interactions.4 Though, orbital interactions are important, too.
2-Fluoroethanol, 2-chloroethanol and their protonated forms
The MP2/6-311++G** geometry optimizations of 2-fluoroethanol (FE) and 2-chloroethanol (CE) led to the five energetically distinguishable minima, shown in Fig. 2 along with their relative electronic energies. They are denoted as aa, ag, ga, gg and gg′, where the first letter refers to the heavy atom F(Cl)–C–C–O conformation, the second to the C–C–O–H conformation. The ag and ag′ conformers are mirror images and energetically indistinguishable. They are both included in discussion to be compared with the corresponding gg and gg′ forms. Optimized geometrical parameters for all conformers of FE and CE are given in Tables S3 and S4 in the ESI.† They compare well with the available experimental data from electron diffraction studies.12a,19a The most stable forms for both FE and CE are gg′ having a hydroxyl hydrogen atom pointing toward the halogen atom in the heavy atom gauche arrangement. The anti conformers of FE are by 2.3 and 2.5 kcal mol−1 higher in energy and this is consistent with previous experimental and theoretical studies.12 The energy difference between gg′ form of CE and anti conformers is smaller and amounts ∼1.7 kcal mol−1, at the employed theory level. This is also in accord with previous experimental and theoretical data.12d,f,g,19 | ||
| Fig. 2 Energy minimum conformations of 2-fluoroethanol (FE) and 2-chloroethanol (CE), their relative electronic energies (kcal mol−1) and energy changes occurring upon anti → gauche rotation of the heavy atom chain. | ||
In the following, the energy change accompanying anti → gauche rotation is analyzed as the energy difference between gauche conformers and their corresponding anti conformers, having the same conformational arrangement around the C–O bonds (anti, + gauche or − gauche, Fig. 2). Decomposition of the total binding energy between two radical fragments into its components is given in Table 3, along with the decomposition of energy change upon anti → gauche rotation. Stabilization energies due to the vicinal total (synclinal and anti) and anti hyperconjugative interactions between the two XCH2 and CH2OH units, evaluated by the NBO analysis, are listed in Table 4, including also energy changes following the anti → gauche conformational isomerization.
| Conformation | ΔEelstat | ΔEex+rep | ΔEpol | ΔEdisp | ΔEint | ΔEprep | ΔE |
|---|---|---|---|---|---|---|---|
| a Labeling of various energy terms is the same as in Table 1.b Structure having the OH hydrogen atom oriented toward a halogen atom. | |||||||
| aa-FE | −150.80 | 221.88 | −154.60 | −27.69 | −111.21 | 17.22 | −93.99 |
| ag-FE | −155.52 | 233.81 | −157.32 | −25.64 | −104.67 | 10.91 | −93.76 |
| ga-FE | −153.53 | 229.18 | −158.42 | −28.11 | −110.88 | 16.74 | −94.14 |
| gg-FE | −156.80 | 237.67 | −159.87 | −26.33 | −105.33 | 11.66 | −93.67 |
| gg′-FEb | −160.86 | 239.58 | −160.35 | −26.15 | −107.78 | 11.48 | −96.30 |
| aa-FE → ga-FE | −2.73 | 7.30 | −3.82 | −0.42 | 0.33 | −0.48 | −0.15 |
| ag-FE → gg-FE | −1.28 | 3.86 | −2.55 | −0.69 | −0.66 | 0.75 | 0.09 |
| ag′-FE → gg′-FE | −5.34 | 5.77 | −3.03 | −0.51 | −3.11 | 0.57 | −2.54 |
| aa-CE | −150.42 | 224.13 | −155.33 | −31.35 | −112.97 | 19.14 | −93.83 |
| ag-CE | −155.07 | 235.40 | −157.47 | −29.33 | −106.47 | 12.61 | −93.86 |
| ga-CE | −151.71 | 229.20 | −157.58 | −31.74 | −111.83 | 18.52 | −93.31 |
| gg-CE | −154.48 | 236.57 | −158.39 | −29.97 | −106.27 | 13.37 | −92.90 |
| gg′-CEb | −158.52 | 239.37 | −159.48 | −29.99 | −108.62 | 13.08 | −95.54 |
| aa-CE → ga-CE | −1.29 | 5.07 | −2.25 | −0.39 | 1.14 | −0.62 | 0.52 |
| ag-CE → gg-CE | 0.59 | 1.17 | −0.92 | −0.64 | 0.20 | 0.76 | 0.96 |
| ag′-CE → gg′-CE | −3.45 | 3.97 | −2.01 | −0.66 | −2.15 | 0.46 | −1.69 |
| FE | CE | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| aa | ag | ga | gg | gg′ | aa | ag | ga | gg | gg′ | |
| Vicinal hyperconjugation | ||||||||||
| E(2)synclinal/anti | 19.33 | 19.20 | 23.06 | 23.79 | 23.14 | 21.84 | 21.59 | 24.67 | 25.28 | 24.98 |
| E(2)synclinal/anti a → g | 3.73 | 4.59 | 3.94 | 2.83 | 3.69 | 3.39 | ||||
| E(del)synclinal/anti | 18.13 | 18.05 | 21.23 | 21.94 | 21.34 | 20.56 | 20.46 | 22.56 | 23.16 | 22.84 |
| E(del)synclinal/anti a → g | 3.10 | 3.88 | 3.28 | 2.00 | 2.70 | 2.38 | ||||
|
||||||||||
| Anti hyperconjugation | ||||||||||
| C–H → C–H* | 2.81 | 2.85 | 2.92 | 2.97 | 3.01 | 2.95 | 2.96 | 3.14 | 3.02 | 3.03 |
| 2.78 | 2.74 | 2.85 | 3.27 | 3.10 | 2.95 | 2.82 | 3.17 | 3.65 | 3.42 | |
| 2.74 | 2.73 | 3.18 | 3.15 | |||||||
| 2.73 | 2.97 | 3.18 | 3.47 | |||||||
| C−X → C–O* | 1.57 | 1.70 | 3.15 | 3.44 | ||||||
| C–O → C–X* | 2.29 | 2.06 | 2.91 | 2.56 | ||||||
| C–H → C–X* | 5.79 | 6.15 | 5.95 | 7.01 | 7.29 | 7.26 | ||||
| C–H → C–O* | 4.63 | 5.51 | 4.89 | 4.36 | 5.28 | 4.85 | ||||
| C–X → C–H* | 0.94 | 0.92 | 1.07 | 2.01 | 1.93 | 2.13 | ||||
| C–O → C–H* | 1.02 | 0.85 | 0.98 | 1.28 | 1.00 | 1.12 | ||||
| Total E(2)anti | 14.92 | 15.05 | 18.15 | 19.67 | 19.00 | 18.32 | 18.40 | 20.97 | 22.17 | 21.81 |
| Total E(2)anti a → g | 3.23 | 4.62 | 3.95 | 2.65 | 3.77 | 3.41 | ||||
The rotation of aa-FE into the corresponding gauche form, that is aa-FE → ga-FE (Fig. 2 and Table 3), results in a very small gauche preference of ΔE = −0.15 kcal mol−1, whereas the ag-FE → gg-FE rotation slightly increases the energy of the system by ΔE = 0.09 kcal mol−1. The true gauche stabilization occurs only upon ag′-FE → gg′-FE rotation, ΔE = −2.54 kcal mol−1. These findings compare with the results of Briggs et al.,12e which led authors to conclude that gauche effect in FE does not have its origin in stereoelectronic effects, but comes from intramolecular hydrogen bonding stabilization. An inspection of data in Tables 3 and 4, however, show that anti hyperconjugative interactions (total E(2)anti a → g in Table 4), all vicinal hyperconjugative interactions (E(2)synclinal/anti a → g and E(del)synclinal/anti a → g in Table 4) and all orbital interactions (ΔEpol in Table 3) become stronger during the all three rotations. Thus, there exists stereoelectronic gauche effect in FE. The most important anti hyperconjugative interactions in gauche structures are C–H → C–F* and C–H → C–O*, the former being more pronounced (Table 4). This arises from a combination of two factors: lower energy of the C–F* orbital and its larger polarization toward the neighbouring carbon atom, due to the larger electronegativity of fluorine atom. The latter allows a stronger orbital overlap.6b The electrostatic energy term ΔEelstat becomes more stabilizing for all three conformational changes, which is also the case for dispersion interactions ΔEdisp, though with smaller magnitude (Table 3). For the aa-FE → ga-FE rotation, these stabilizing interactions are not large enough to override a destabilization coming from the increase in the steric repulsion (which is the strongest in this case), thus making the ΔEint positive (0.33 kcal mol−1). The slight gauche preference for this rotation actually comes from an energy loss due to the accompanying structural changes, ΔEprep = −0.48 kcal mol−1. In the case of ag-FE → gg-FE conformational change, the interaction energy becomes more stabilizing, ΔEint = −0.66 kcal mol−1, but not enough to overcome the increase in the ΔEprep = 0.75 kcal mol−1 and there is almost no conformational preference for this rotation. The pronounced gauche stabilization for ag′-FE → gg′-FE rotation of ΔE = −2.54 kcal mol−1 results from a favourable interaction energy, ΔEint = −3.11 kcal mol−1, while ΔEprep rises by 0.57 kcal mol−1. The interaction energy stabilization owes mainly to the electrostatic energy ΔEelstat = −5.34 kcal mol−1, followed by the orbital interaction energy ΔEpol = −3.03 kcal mol−1. The dispersion energy contribution is significantly smaller, ΔEdisp = −0.51 kcal mol−1 (Table 3). Hence, even in the neutral FE, the most important contributor to the gauche effect is electrostatic energy. A part of electrostatic stabilization should be ascribed to the attraction between the two antiparallel C–F and O–H bond dipoles, with θFCOH = 4.3°. However, other electrostatic stabilizing interactions must be involved, too, since ΔEelstat becomes more stabilizing even for the remaining two rotations which bring fluorine atom near the oxygen lone pair. In these cases, an electrostatic repulsion between the C–F and C–O bond dipoles and between fluorine and oxygen lone pairs is anticipated. Our results, however, once again point out that partial interaction between bond pairs and lone pairs is not always enough to account for the overall change in electrostatic interactions.
Now, let us examine what happens when fluorine atom is replaced with the less electronegative and larger chlorine atom. Both aa-CE → ga-CE and ag-CE → gg-CE rotations are followed by an increase in energy by ΔE = 0.52 and ΔE = 0.96 kcal mol−1, respectively, which is due to the ΔEint = 1.14 kcal mol−1 in the first case, ΔEint = 0.20 kcal mol−1 and ΔEprep = 0.76 kcal mol−1 in the second case. The favourable preparation energy for the former isomerization reduces the interaction energy rise by ΔEprep = −0.62 kcal mol−1 (Table 3). The ag′-CE → gg′-CE rotation still leads to gauche preference, though smaller than in FE (ΔE = −1.69 kcal mol−1 for CE and ΔE = −2.54 kcal mol−1 for FE, Fig. 2 and Table 3). This is due to the ΔEint = −2.15 kcal mol−1, while ΔEprep increases the energy by 0.46 kcal mol−1 (and is smaller compared to the corresponding rotation of FE, with ΔEprep = 0.57 kcal mol−1).
An examination of data in Table 3 enables us to explain the observed changes in ΔEint during the three rotations of CE and its smaller gauche effect (ag′-CE → gg′-CE isomerization) compared to that in FE. Similarly as in the case of CFE, the interaction energy rise (positive ΔEint for the first two rotations) and its smaller negative change for the third rotation do not have their origin in the steric effect. The increase in the ΔEex+rep is smaller for CE than for FE for all three rotations. Again, electrostatic and orbital stabilizing interactions are smaller for the anti → gauche rotation in CE compared to FE (change in ΔEelstat is even destabilizing (positive) for the ag-CE → gg-CE rotation). This decrease in the ΔEelstat and ΔEpol makes the ΔEint energy term positive for the aa-CE → ga-CE and ag-CE → gg-CE conformational changes, and less negative (by ∼1 kcal mol−1) for the ag′-CE → gg′-CE isomerization relative to the ag′-FE → gg′-FE one. For all three rotations, dispersion energies are favourable, though their stabilizing effect does not exceed 0.7 kcal mol−1. Thus, as in the case of CFE, the less electronegative and larger chlorine atom provides smaller electrostatic and orbital stabilization to the anti → gauche conformational change, compared to fluoroalcohol.
A part of a decrease in the orbital interaction energy can be analyzed by examination of the NBO results shown in Table 4. Total and anti vicinal hyperconjugative stabilization associated with the anti → gauche isomerization is smaller in CE than in FE. This should be ascribed to larger increase in hyperconjugation in anti forms of CE than in gauche forms relative to FE. The increase in the hyperconjugative stabilization in anti conformers of CE mainly stems from the more favourable C–Cl → C–O* interaction (3.2–3.4 kcal mol−1) vs. C–F → C–O* interaction (1.6–1.7 kcal mol−1) in FE, consistent with the better electron-donating ability of the C–Cl bond, as already mentioned above. There are also contributions from other interactions, but smaller in magnitude. In the case of gauche conformers, main contribution to larger hyperconjugative stabilization comes from an increase in the C–H → C–Cl* (7–7.3 kcal mol−1) and C–Cl → C–H* interactions (1.9–2.1 kcal mol−1) in CE vs. C–H → C–F* (5.8–6.2 kcal mol−1) and C–F → C–H* (0.9–1.1 kcal mol−1) in FE, and this compares with the results for CFE vs. DFE (Tables 2 and 4). Thus, the anti → gauche isomerization benefits less from hyperconjugation in the case of CE compared to FE.
Overall, the gauche effect in CE, observed for the ag′-CE → gg′-CE isomerization, has to be ascribed to electrostatic energy as the main stabilizing factor (ΔEelstat = −3.45 kcal mol−1), orbital interaction energy (ΔEpol = −2.01 kcal mol−1) and dispersion energy (ΔEdisp = −0.66 kcal mol−1). In this case, too, a part of electrostatic stabilization should be attributed to the attraction between the nearly parallel, but oppositely oriented C–Cl and O–H bond dipoles, with θClCOH = 3°.
The protonated FE and CE, abbreviated herein as FEH and CEH, respectively, can exist as five energetically distinguishable conformers denoted as aa, ag, ga, gg and gg′, where the first letter refers to the heavy atom F(Cl)–C–C–O conformation, the second to the C–C–O–: conformation They are shown in Fig. 3, along with their relative energies. The ag′ conformer is enantiomeric with the ag one, and is included in the discussion for comparison with the corresponding gg′ conformer. As with the neutral alcohols, energies of gauche conformers are analyzed with respect to their corresponding anti forms (Fig. 3). The optimized structural parameters for FEH and CEH are given in Tables S5 and S6 in the ESI.†
 | ||
| Fig. 3 Energy minimum conformations of protonated 2-fluoroethanol (FEH) and protonated 2-chloroethanol (CEH), their relative electronic energies (kcal mol−1) and energy changes occurring upon anti → gauche rotation of the heavy atom chain. | ||
The protonated FE shows very large gauche preference for all three rotations (aa-FEH → ga-FEH, ag-FEH → gg-FEH, ag′-FEH → gg′-FEH), particularly pronounced when gauche forms have a hydroxyl hydrogen atom oriented toward the fluorine atom (the first two rotations in Fig. 3). These findings are consistent with previous study on protonated FE,12e which led authors to conclude that in this case the gauche effect originates from both intramolecular hydrogen bonding stabilization and stereoelectronic effects. The latter was seen as the only stabilization in the ag′-FEH → gg′-FEH rotation, since there is no hydrogen bond in the gg′ conformation.12e Our energy decomposition and NBO data reveal that all orbital stabilization (ΔEpol in Table 5), as well as anti and total vicinal hyperconjugation (Table 6) are indeed larger in gauche conformations by 5.3–5.8 kcal mol−1, 3.3–6.4 kcal mol−1 and 3.7–6.7 kcal mol−1, respectively. Among the anti hyperconjugative interactions, the most important one contributing to the stabilization of the gauche forms is the C–H → C–O* interaction, having stabilizing energy of 6.4–9 kcal mol−1. The C–H → C–F* interaction contributes smaller stabilizing energy of 3.5–4.1 kcal mol−1. Thus, protonation of hydroxy group enhances electron-accepting ability of the C–O* antibond and reverses the relative strength of C–H → C–F* and C–H → C–O* interactions in neutral and protonated alcohols. This enhancement arises from the C–O* antibond energy lowering and larger polarization. Our conclusions, thus, agree with those of Briggs et al.12e that there exists stereoelectronic gauche effect in protonated FE.
| Conformation | ΔEelstat | ΔEex+rep | ΔEpol | ΔEdisp | ΔEint | ΔEprep | ΔE |
|---|---|---|---|---|---|---|---|
| a Labeling of various energy terms is the same as in Table 1.b Structure having an OH2+ hydrogen atom oriented toward a halogen atom. | |||||||
| aa-FEH | −128.96 | 202.24 | −154.75 | −28.01 | −109.48 | 9.46 | −100.02 |
| ag-FEH | −128.21 | 198.90 | −154.83 | −28.43 | −112.57 | 12.03 | −100.54 |
| ga-FEHb | −141.80 | 211.84 | −160.01 | −28.17 | −118.14 | 10.27 | −107.87 |
| gg-FEHb | −140.53 | 208.42 | −160.23 | −29.27 | −121.61 | 12.84 | −108.77 |
| gg′-FEH | −135.09 | 207.31 | −160.65 | −29.15 | −117.58 | 12.83 | −104.75 |
| aa-FEH → ga-FEH | −12.84 | 9.60 | −5.26 | −0.16 | −8.66 | 0.80 | −7.86 |
| ag-FEH → gg-FEH | −12.32 | 9.52 | −5.40 | −0.84 | −9.04 | 0.81 | −8.23 |
| ag′-FEH → gg′-FEH | −6.88 | 8.41 | −5.82 | −0.72 | −5.01 | 0.80 | −4.21 |
| aa-CEH | −132.52 | 212.11 | −162.00 | −32.47 | −114.88 | 11.86 | −103.02 |
| ag-CEH | −131.86 | 209.34 | −162.31 | −33.04 | −117.87 | 14.53 | −103.34 |
| ga-CEHb | −142.38 | 221.24 | −166.33 | −34.33 | −121.80 | 12.82 | −108.98 |
| gg-CEHb | −141.34 | 218.99 | −167.35 | −35.65 | −125.35 | 15.32 | −110.03 |
| gg′-CEH | −134.52 | 211.04 | −163.05 | −33.85 | −120.38 | 15.26 | −105.12 |
| aa-CEH → ga-CEH | −9.86 | 9.13 | −4.33 | −1.86 | −6.92 | 0.96 | −5.96 |
| ag-CEH → gg-CEH | −9.48 | 9.65 | −5.04 | −2.61 | −7.48 | 0.79 | −6.69 |
| ag′-CEH → gg′-CEH | −2.66 | 1.70 | −0.74 | −0.81 | −2.51 | 0.72 | −1.79 |
| FEH | CEH | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| aa | ag | ga | gg | gg′ | aa | ag | ga | gg | gg′ | |
| Vicinal hyperconjugation | ||||||||||
| E(2)synclinal/anti | 18.64 | 18.48 | 22.77 | 22.17 | 25.23 | 24.99 | 24.77 | 25.06 | 24.56 | 27.67 |
| E(2)synclinal/anti a → g | 4.13 | 3.69 | 6.75 | 0.07 | −0.21 | 2.90 | ||||
| E(del)synclinal/anti | 17.63 | 17.40 | 20.76 | 20.19 | 22.73 | 23.26 | 22.95 | 22.87 | 22.36 | 24.93 |
| E(del)synclinal/anti a → g | 3.13 | 2.80 | 5.33 | −0.39 | −0.59 | 1.97 | ||||
|
||||||||||
| Anti hyperconjugation | ||||||||||
| C–H → C–H* | 2.74 | 2.67 | 2.83 | 2.88 | 2.94 | 3.10 | 3.03 | 2.80 | 2.92 | 3.18 |
| 2.74 | 2.76 | 2.45 | 2.21 | 2.47 | 3.10 | 3.16 | 2.89 | 2.64 | 2.89 | |
| 2.10 | 2.07 | 2.70 | 2.73 | |||||||
| 2.10 | 1.87 | 2.70 | 2.43 | |||||||
| C–X → C–O* | 3.44 | 3.20 | 8.46 | 7.99 | ||||||
| C–O → C–X* | 1.22 | 1.34 | 1.55 | 1.72 | ||||||
| C–H → C–X* | 3.53 | 3.66 | 4.08 | 4.55 | 4.75 | 5.12 | ||||
| C–H → C–O* | 7.22 | 6.37 | 8.97 | 7.45 | 6.66 | 9.33 | ||||
| C–X → C–H* | 1.22 | 1.17 | 1.04 | 2.47 | 2.36 | 2.22 | ||||
| C–O → C–H* | 0.87 | 0.95 | 0.83 | 1.00 | 1.13 | 1.03 | ||||
| Total E(2)anti | 14.34 | 13.91 | 18.12 | 17.24 | 20.33 | 21.61 | 21.06 | 21.16 | 20.46 | 23.77 |
| Total E(2)anti a → g | 3.78 | 3.33 | 6.42 | −0.45 | −0.60 | 2.71 | ||||
Energy decomposition data, given in Table 5, reveal which other factors contribute to the very strong gauche preference in FEH. Thus, in all three isomerizations it results from ΔEint, while ΔEprep increases by 0.8 kcal mol−1. Major factor which is responsible for the large gauche conformer stabilization in the aa-FEH → ga-FEH and ag-FEH → gg-FEH rotations is a very favourable electrostatic energy component, which reaches values of 12.84 kcal mol−1 and 12.32 kcal mol−1, respectively (ΔEelstat in Table 5). Electrostatic stabilization energy is smaller, but still significant for the ag′-FEH → gg′-FEH conformational change, 6.88 kcal mol−1. In the first two cases, the large electrostatic stabilization partly arises from an attraction between the two almost antiparallel O–H and C–F bond dipoles, having θFCOH 6.4° and 8.1°, respectively. There are, however, other attractive contributions, since the electrostatic energy becomes more favourable even for the third rotation, when the gauche form is expected to be destabilized by the repulsion between the two C–O and C–F bond dipoles. The calculated NBO charges showed that the positive charge in the C–OH2+ fragment is distributed on hydrogen and carbon atoms whereas the oxygen contains the negative charge. In addition, electrostatic repulsion between lone pairs should be involved, too.
To conclude, the ΔEelstat energy component provides the most important contribution to the gauche effect in FEH. Next comes the orbital interaction energy, which ranges from 5.2–5.8 kcal mol−1. They both, along with the much smaller ΔEdisp, 0.2–0.8 kcal mol−1 make the ΔEint very favourable, by strongly overcoming the also prominent increase in the steric repulsion (8.4–9.6 kcal mol−1).
Our extension to chloro derivatives shows that protonation of CE also leads to the pronounced gauche preference of 6–6.7 kcal mol−1 (Fig. 3 and Table 5) compared to only 1.7 kcal mol−1 in the neutral alcohol (Fig. 2 and Table 3), taking into account conformational changes leading to the gauche structures having a hydroxyl hydrogen atom oriented toward the chlorine atom (aa-CEH → ga-CEH and ag-CEH → gg-CEH rotations for the protonated alcohol, and ag′-CE → gg′-CE rotation for the alcohol). Contrary to the neutral CE, the protonated molecule also shows gauche stabilization for the ag′-CEH → gg′-CEH rotation when chlorine atom becomes oriented toward the oxygen lone pair. The energy of this stabilization is smaller and amounts ΔE = −1.79 kcal mol−1. For all three rotations, depicted in Fig. 3, it is the ΔEint that favours the conformational change (2.5–7.5 kcal mol−1), whereas structural changes lead to an increase in energy by 0.7–1 kcal mol−1 (Table 5). The most important factor that makes ΔEint more negative is ΔEelstat, for all three rotations. There is a strong electrostatic stabilization for the aa-CEH → ga-CEH and ag-CEH → gg-CEH conformational changes which amounts 9.86 kcal mol−1 and 9.48 kcal mol−1, respectively, and the smaller one for the ag′-CEH → gg′-CEH rotation of 2.66 kcal mol−1, but still stabilizing despite the expected lone pairs repulsion and C–Cl/C–O bond dipoles repulsion. As with the fluoro derivative, the oxygen atom is the negative part of the C–O bond dipole in the C–OH2+ unit. The next stabilizing effect comes from orbital interactions and dispersion energy term which is by ∼1.7 kcal mol−1 stronger than in FEH, for the first two isomerizations. The ΔEpol for the aa-CEH → ga-CEH (4.33 kcal mol−1) and ag-CEH → gg-CEH (5.04 kcal mol−1) rotations is significantly greater than that for the ag′-CEH → gg′-CEH rotation (0.74 kcal mol−1), while the NBO results show that there is no hyperconjugative stabilization following the first two rotations (Table 6), which should be ascribed mainly to strong C–Cl → C–O* interaction in anti conformers (8–8.5 kcal mol−1, compared to 3.2–3.4 kcal mol−1 for the C–F → C–O* interaction in FEH). The third isomerization is the only one that has stereoelectronic origin, in addition to other orbital effects. Thus, the more favourable ΔEpol component observed for the aa-CEH → ga-CEH and ag-CEH → gg-CEH rotations, which compares with the corresponding values for FEH, should be related to polarization effects and charge transfer in hydrogen bonding interactions. Indeed, the NBO(del) calculations revealed a very large charge transfer component of hydrogen bonding in ga-CEH and gg-CEH, which amount 10.6 kcal mol−1 and 13.3 kcal mol−1, respectively. This has to be compared with only 1.2 kcal mol−1 and 2 kcal mol−1 for ga-FEH and gg-FEH, 0.6 kcal mol−1 for gg′-CE and 0.2 kcal mol−1 for gg′-FE. Though, it should be noted that NBO overestimates charge transfer interactions compared to the BLW method,28,40 but their trend is the same.39
The steric strain arising from the closer contact between Cl and OH2+ substituents in gauche conformers is very similar to that observed for FEH regarding the aa-CEH → ga-CEH and ag-CEH → gg-CEH rotations, and significantly smaller for the ag′-CEH → gg′-CEH change (Table 5). This means that the decreased gauche effect in CEH compared to that in FEH does not originate from steric effects, but from a decrease in electrostatic stabilizing energy, which is 12.3–12.8 kcal mol−1 for the first two rotations in FEH, but 9.5–9.9 kcal mol−1 for the same conformational changes in CEH. There is a drop in this energy term for the third ag′-CEH → gg′-CEH rotation, as well (6.9 kcal mol−1 for FEH and 2.7 kcal mol−1 for CEH). A drop in the orbital interactions is less pronounced except for the ag′-CEH → gg′-CEH rotation, lacking the intramolecular hydrogen bond (Table 5). Therefore, only for the ag′ → gg′ conformational change the stronger gauche preference in FEH owes to both electrostatic and orbital stabilizing interactions. In the case of the other two aa → ga and ag → gg isomerizations, the larger gauche effect in FEH originates mostly from more favourable electrostatic energy.
2-Fluoroethylamine, 2-chloroethylamine and their protonated forms
The MP2/6-311++G** geometry optimizations of 2-fluoroethylamine (FEA) and 2-chloroethylamine (CEA) led to the five energetically distinguishable minima, depicted in Fig. 4 along with their relative electronic energies. They are denoted as aa, ag, ga, gg and gg′, where the first letter refers to the heavy atom F(Cl)–C–C–N conformation and the second to the C–C–N–: conformation. Here, again, the enantiomeric ag and ag′ conformers are both included to be compared with the corresponding gg and gg′ forms. Optimized geometrical parameters for all conformers of FEA and CEA are given in Tables S7 and S8 in the ESI.† For FEA, they are in good agreement with the available experimental data.13d The two most stable conformers for both FEA and CEA are those having one of amino hydrogen atoms pointing toward the halogen atom in the heavy atom gauche arrangement. Among them, the gg form is slightly more stable than the ga form. In the case of FEA, this agrees with previous experimental13a,d and theoretical studies.13c The relative energies of five conformers of FEA are also in accord with the existing experimental data,13d though the energy difference between the aa and ag forms is small (0.12 kcal mol−1) and their relative stability may vary with the level of computations.13c The same applies to CEA, for which aa and ag conformers differ by 0.17 kcal mol−1 in our calculations. Conformers ga-CEA and gg-CEA also have similar energies which is in accord with previous experimental and theoretical data,20 although their relative stability order is reversed in our MP2 calculations. The relative energies for other three conformers of CEA are in agreement with the exisiting data.20 As in the case of 2-haloalcohols, the gauche preference in amines is smaller when halogen is chlorine atom.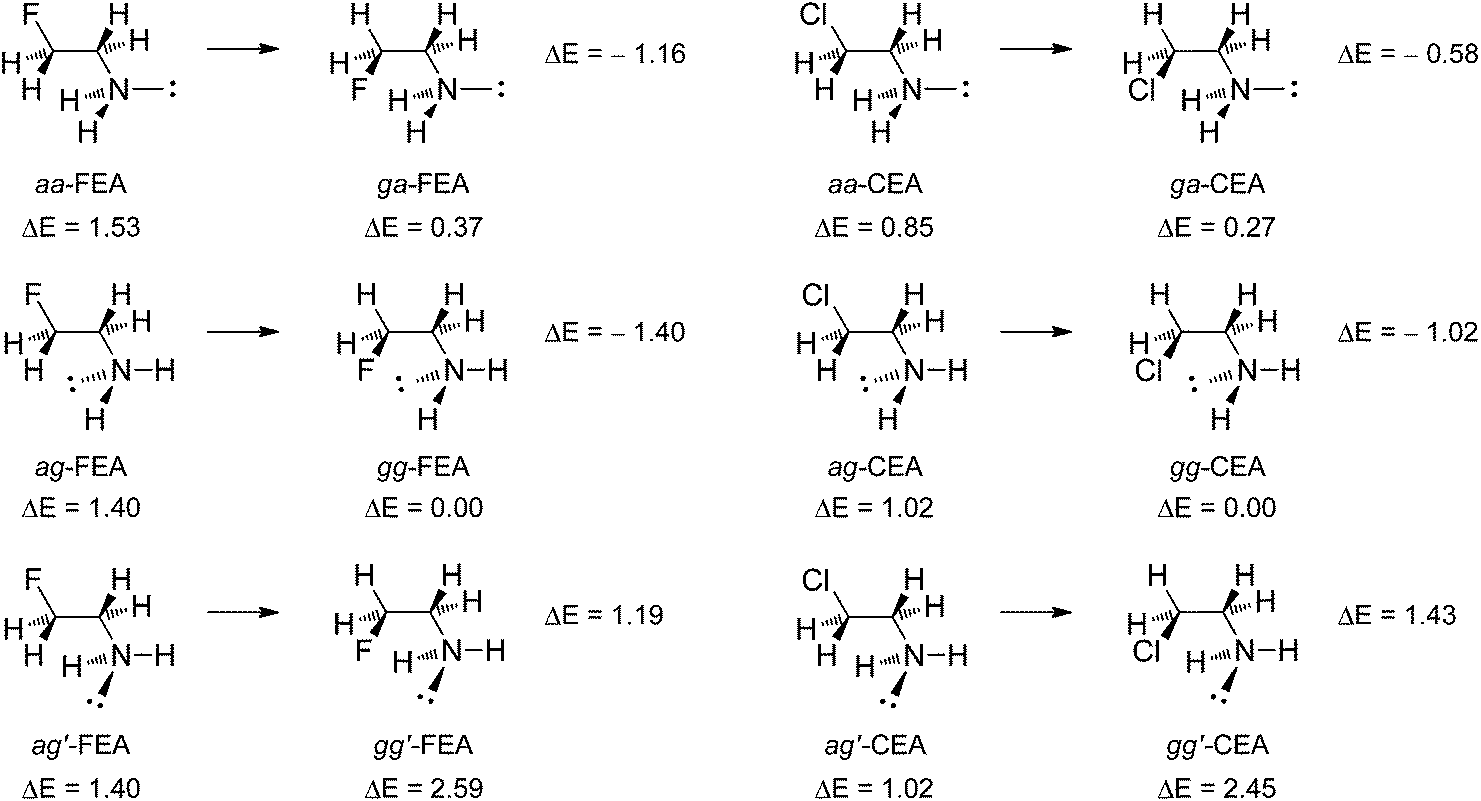 | ||
| Fig. 4 Energy minimum conformations of 2-fluoroethylamine (FEA) and 2-chloroethylamine (CEA), their relative electronic energies (kcal mol−1) and energy changes occurring upon anti → gauche rotation of the heavy atom chain. | ||
Decomposition of the total binding energy between the two XCH2˙ and ˙CH2NH2 radical fragments into its components is given in Table 7, along with the decomposition of energy change occurring upon anti → gauche rotation of the heavy atom chain. Stabilization energies due to the vicinal anti hyperconjugative interactions and all vicinal hyperconjugative interactions (synclinal and anti) between the two XCH2 and CH2NH2 units, evaluated by the NBO analysis, are listed in Table 8, including energy changes following anti → gauche conformational isomerization (denoted as a → g).
| Conformation | ΔEelstat | ΔEex+rep | ΔEpol | ΔEdisp | ΔEint | ΔEprep | ΔE |
|---|---|---|---|---|---|---|---|
| a Labeling of various energy terms is the same as in Table 1.b Structure having an NH2 hydrogen atom oriented toward a halogen atom. | |||||||
| aa-FEA | −160.85 | 243.68 | −159.29 | −25.81 | −102.27 | 11.49 | −90.78 |
| ag-FEA | −155.31 | 230.10 | −155.91 | −27.54 | −108.66 | 17.74 | −90.92 |
| ga-FEAb | −163.56 | 246.11 | −160.46 | −25.87 | −103.78 | 11.83 | −91.95 |
| gg-FEAb | −156.77 | 231.14 | −157.53 | −28.82 | −111.98 | 19.67 | −92.31 |
| gg′-FEA | −154.09 | 231.18 | −157.17 | −28.23 | −108.31 | 18.59 | −89.72 |
| aa-FEA → ga-FEA | −2.71 | 2.43 | −1.17 | −0.06 | −1.51 | 0.34 | −1.17 |
| ag-FEA → gg-FEA | −1.46 | 1.04 | −1.62 | −1.28 | −3.32 | 1.93 | −1.39 |
| ag′-FEA → gg′-FEA | 1.22 | 1.08 | −1.26 | −0.69 | 0.35 | 0.84 | 1.19 |
| aa-CEA | −159.58 | 243.59 | −158.42 | −29.51 | −103.92 | 13.18 | −90.74 |
| ag-CEA | −154.10 | 230.80 | −155.67 | −31.34 | −110.31 | 19.74 | −90.57 |
| ga-CEAb | −160.79 | 244.63 | −158.81 | −29.74 | −104.71 | 13.39 | −91.32 |
| gg-CEAb | −154.99 | 231.76 | −156.92 | −32.75 | −112.90 | 21.32 | −91.58 |
| gg′-CEA | −152.16 | 230.71 | −156.07 | −32.07 | −109.59 | 20.46 | −89.13 |
| aa-CEA → ga-CEA | −1.21 | 1.04 | −0.39 | −0.23 | −0.79 | 0.21 | −0.58 |
| ag-CEA → gg-CEA | −0.89 | 0.96 | −1.25 | −1.41 | −2.59 | 1.58 | −1.01 |
| ag′-CEA → gg′-CEA | 1.94 | −0.09 | −0.40 | −0.73 | 0.72 | 0.72 | 1.44 |
| FEA | CEA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| aa | ag | ga | gg | gg′ | aa | ag | ga | gg | gg′ | |
| Vicinal hyperconjugation | ||||||||||
| E(2)synclinal/anti | 20.02 | 20.03 | 23.11 | 22.80 | 22.79 | 22.04 | 22.25 | 24.98 | 24.85 | 24.66 |
| E(2)synclinal/anti a → g | 3.09 | 2.77 | 2.76 | 2.94 | 2.60 | 2.41 | ||||
| E(del)synclinal/anti | 18.84 | 18.85 | 21.36 | 21.04 | 21.06 | 20.86 | 20.95 | 22.90 | 22.73 | 22.62 |
| E(del)synclinal/anti a → g | 2.52 | 2.19 | 2.21 | 2.03 | 1.78 | 1.67 | ||||
|
||||||||||
| Anti hyperconjugation | ||||||||||
| C–H → C–H* | 3.03 | 2.99 | 3.14 | 3.16 | 3.06 | 3.08 | 3.11 | 3.13 | 3.21 | 3.21 |
| 3.03 | 2.99 | 3.38 | 3.14 | 3.23 | 3.08 | 3.07 | 3.71 | 3.42 | 3.56 | |
| 3.12 | 2.80 | 3.54 | 3.22 | |||||||
| 3.12 | 3.11 | 3.54 | 3.58 | |||||||
| C–X → C–N* | 1.68 | 1.50 | 3.27 | 3.03 | ||||||
| C–N → C–X* | 2.32 | 2.68 | 2.93 | 3.40 | ||||||
| C–H → C–X* | 6.09 | 6.17 | 5.93 | 7.45 | 7.46 | 7.23 | ||||
| C–H → C–N* | 4.92 | 4.06 | 4.43 | 4.91 | 4.02 | 4.24 | ||||
| C–X → C–H* | 1.10 | 1.13 | 0.98 | 2.04 | 2.25 | 2.04 | ||||
| C–N → C–H* | 1.03 | 1.26 | 1.08 | 1.11 | 1.46 | 1.29 | ||||
| Total E(2)anti | 16.30 | 16.07 | 19.66 | 18.92 | 18.71 | 19.44 | 19.41 | 22.35 | 21.82 | 21.57 |
| Total E(2)anti a → g | 3.36 | 2.85 | 2.64 | 2.91 | 2.41 | 2.16 | ||||
In the case of FEA, the aa-FEA → ga-FEA and ag-FEA → gg-FEA conformational isomerizations lead to a decrease in energy (stabilization of the system) by ΔE = −1.16 kcal mol−1 and ΔE = −1.40 kcal mol−1, respectively. This has to be ascribed to the ΔEint, while ΔEprep increases by 0.34 kcal mol−1 and 1.93 kcal mol−1, respectively. For the first rotation, ΔEelstat = −2.71 kcal mol−1 is more stabilizing than ΔEpol = −1.17 kcal mol−1, while dispersion energy contribution is negligible (Table 7). For the second rotation, ΔEpol = −1.62 kcal mol−1 is slightly more favourable than ΔEelstat = −1.46 kcal mol−1, and the dispersion energy stabilizing contribution increases to ΔEdisp = −0.69 kcal mol−1. In both cases, these stabilizing interactions overcome the increase in the steric repulsion, introduced mostly by bringing the two substituents into the gauche position. Thus, the gauche preference in FEA should be ascribed to both electrostatic and orbital stabilizing interactions. Data in Table 8 show that vicinal hyperconjugative interactions between the two FCH2 and CH2NH2 fragments also become stabilizing upon aa-FEA → ga-FEA and ag-FEA → gg-FEA isomerizations, meaning that stereoelectronic effects contribute to the gauche stabilization, too. The most important hyperconjugative interactions that stabilize the gauche forms are C–H → C–F* (∼6 kcal mol−1) and C–H → C–N* (4.1–4.9 kcal mol−1).
The third ag′-FEA → gg′-FEA rotation does not result in molecule stabilization, that is it increases the energy by ΔE = 1.19 kcal mol−1, both because of an increase in ΔEint and ΔEprep (Fig. 4 and Table 7). The stabilizing stereoelectronic effects, as well as all orbital stabilizing interactions are still present, as evidenced from the data in Tables 7 (ΔEpol) and 8. The increase in the steric repulsion does not exceed values for the first two isomerizations. What is missing now is more stabilizing electrostatic interaction, since ΔEelstat becomes less favourable (positive) by 1.22 kcal mol−1. This differs from the case of FE, when electrostatic energy favours gauche conformation even when fluorine atom encounters oxygen lone pairs (compare data for aa-FE → ga-FE and ag-FE → gg-FE rotations in Table 3 with those for ag′-FEA → gg′-FEA rotation in Table 7). This reinforces our above mentioned conclusion that the favourable electrostatic energy change is more important for smaller and more electronegative atoms. Though, in this case van der Waals radii do not differ much (1.55 Å for N, 1.52 Å for O),21 but electronegativity between N and O atoms differs by 0.5 according to the Pauling electronegativity scale.
The conformational behaviour of CEA resembles the behavior of FEA in that the aa-CEA → ga-CEA and ag-CEA → gg-CEA isomerizations are followed by a decrease in energy by ΔE = −0.58 kcal mol−1 and ΔE = −1.02 kcal mol−1, respectively, whereas the ag′-CEA → gg′-CEA rotation results in an increase in energy by ΔE = 1.43 kcal mol−1 (Fig. 4 and Table 7). In all three cases ΔEprep becomes more positive, so that gauche stabilization in the first two isomerizations results from the more favourable ΔEint. In the latter case, ΔEint increases, as well. The energetic stabilization of the system (gauche preference) is smaller than in FEA, while energetic destabilization following the ag′-CEA → gg′-CEA rotation is larger than in FEA. The reason for this lies in the ΔEint energy, since increase in ΔEprep is smaller than in FEA. The aa-CEA → ga-CEA isomerization owes more to the electrostatic stabilization, ΔEelstat = −1.21 kcal mol−1, than to orbital and dispersion energies, ΔEpol = −0.39 kcal mol−1 and ΔEdisp = −0.23 kcal mol−1. For the ag-CEA → gg-CEA isomerization, both ΔEpol = −1.25 kcal mol−1 and ΔEdisp = −1.41 kcal mol−1 energies are more favourable than ΔEelstat = −0.89 kcal mol−1. This compares with the situation in FEA, with the absolute values of ΔEelstat and ΔEpol being smaller and those of ΔEdisp larger relative to the corresponding values for FEA, which is in accord with the above discussion. In the case of the ag′-CEA → gg′-CEA rotation, the more favourable ΔEpol and ΔEdisp energies are not large enough to override the 1.94 kcal mol−1 smaller electrostatic stabilization in gg′ vs. ag′, resulting in positive ΔEint energy. This electrostatic destabilization is larger compared to the one in FEA (1.22 kcal mol−1) which is due to the replacement of fluorine with chlorine atom. It is also larger than electrostatic destabilization following the ag-CE → gg-CE isomerization (0.59 kcal mol−1, Table 3), which brings chlorine close to the oxygen lone pair, while the aa-CE → ga-CE rotation, also leading to the chlorine/oxygen lone pair interaction results in a more favourable ΔEelstat = −1.29 kcal mol−1. The latter results could be attributed to the already mentioned observation that the more electronegative oxygen atom is more effective in electrostatic stabilization in gauche relative to the anti form, or its smaller destabilization. Interestingly, a change in the steric repulsion accompanying the ag′-CEA → gg′-CEA conformational isomerization is negligible (Table 7).
Among the orbital interactions, vicinal anti and total hyperconjugation do favour heavy atoms gauche arrangement more than the anti, but less so than in FEA (compare a → g values for FEA and CEA in Table 8). The C–H → C–Cl* and C–Cl → C–H* interactions are increased in ga and gg forms of CEA relative to the corresponding interactions in FEA by ∼1.3 kcal mol−1 and ∼1 kcal mol−1, respectively, but concomitantly the C–Cl → C–N* and C–N → C–Cl* interactions become larger in aa and ag conformers of CEA by ∼1.6 kcal mol−1 and ∼0.6–0.7 kcal mol−1, respectively, compared to the corresponding interactions in FEA. Together with somewhat smaller changes in other hyperconjugative interactions, the net result is less pronounced stereoelectronic stabilization for the anti → gauche isomerizations in the case of CEA.
The protonated forms of FEA and CEA, denoted herein as FEAH and CEAH, respectively, exist as one gauche and one anti conformer, which are energetically distinguishable (Fig. 5). The MP2/6-311++G** optimized structural parameters are given in Tables S9 and S10 in the ESI.† The FEAH shows a significant gauche effect of ΔE = −6.85 kcal mol−1 (this work), which was earlier ascribed to a combination of intramolecular hydrogen bonding and stereoelectronic effects,12e,13c or electrostatic interactions.10 Our data from energy decomposition analysis show that this gauche preference originates mostly from electrostatic energy term which is 2.7 times stronger than all orbital interaction stabilization (Table 9). It certainly involves C–F and N–H bond dipoles attraction, which are almost fully parallel (θFCNH = 0.2°) and oppositely oriented. Contribution of ΔEdisp is small. The ΔEelstat and ΔEpol significantly overcome the steric repulsion, thus making the ΔEint very favourable (−7.18 kcal mol−1). It is just slightly diminished by the positive ΔEprep energy. The existence of stereoelectronic gauche effect, too, is evident from stronger vicinal hyperconjugative interactions in g-FEAH with respect to a-FEAH, shown in Table 10. The most important contribution in the gauche form comes from the C–H → C–N* interaction which exceeds the C–H → C–F* one (5.3 kcal mol−1 and 3.9 kcal mol−1, respectively). This contrasts their order in the free amine (Table 8) and is in accord with the relative order of the corresponding interactions in neutral and protonated alcohols.
 | ||
| Fig. 5 Energy minimum conformations of protonated 2-fluoroethylamine (FEAH) and protonated 2-chloroethylamine (CEAH), their relative electronic energies (kcal mol−1) and energy changes occurring upon anti → gauche rotation. | ||
| Conformation | ΔEelstat | ΔEex+rep | ΔEpol | ΔEdisp | ΔEint | ΔEprep | ΔE |
|---|---|---|---|---|---|---|---|
| a Labeling of various energy terms is the same as in Table 1. | |||||||
| a-FEAH | −131.03 | 202.24 | −151.97 | −29.64 | −110.40 | 11.22 | −99.18 |
| g-FEAH | −140.48 | 208.20 | −155.52 | −29.78 | −117.58 | 11.56 | −106.02 |
| a-FEAH → g-FEAH | −9.45 | 5.96 | −3.55 | −0.14 | −7.18 | 0.34 | −6.84 |
| a-CEAH | −132.93 | 208.69 | −156.12 | −33.81 | −114.17 | 13.40 | −100.77 |
| g-CEAH | −139.58 | 213.18 | −158.46 | −34.74 | −119.60 | 13.58 | −106.02 |
| a-CEAH → g-CEAH | −6.65 | 4.49 | −2.34 | −0.93 | −5.43 | 0.19 | −5.24 |
| a-FEAH | g-FEAH | a-CEAH | g-CEAH | |
|---|---|---|---|---|
| Vicinal hyperconjugation | ||||
| E(2)synclinal/anti | 18.59 | 21.04 | 22.38 | 24.22 |
| E(2)synclinal/anti a → g | 2.45 | 1.84 | ||
| E(del)synclinal/anti | 17.56 | 19.50 | 21.12 | 22.31 |
| E(del)synclinal/anti a → g | 1.94 | 1.20 | ||
|
||||
| Anti hyperconjugation | ||||
| C–H → C–H* | 2.88 (×2) | 2.99 | 3.17 (×2) | 3.08 |
| 2.30 (×2) | 2.50 | 2.79 (×2) | 2.95 | |
| C–X → C–N* | 2.42 | 5.26 | ||
| C–N → C–X* | 1.43 | 1.98 | ||
| C–H → C–X* | 3.97 | 5.21 | ||
| C–H → C–N* | 5.31 | 6.07 | ||
| C–X → C–H* | 1.23 | 2.51 | ||
| C–N → C–H* | 0.90 | 1.05 | ||
| Total E(2)anti | 14.21 | 16.90 | 19.16 | 20.87 |
| Total E(2)anti a → g | 2.69 | 1.71 | ||
Protonation of CEA increases the gauche preference from 0.6–1 kcal mol−1 in the free amine (Fig. 4 and Table 7) to 5.24 kcal mol−1 in CEAH (Fig. 5 and Table 9). Compared to CEA, the most important contributor to the increase in the gauche effect has to be attributed to the ΔEelstat energy component, the stabilization of which increases from 0.9–1.2 kcal mol−1 in amine to 6.6 kcal mol−1 in its protonated form. In both compounds, favourable electrostatic energy also involves attraction between the almost antiparallel C–Cl and N–H dipoles, θClCNH being 2.5°, 1.5° and 2.7° in ga-CEA, gg-CEA and g-CEAH, respectively. The second favourable energy contribution in CEAH comes from ΔEpol energy term, which rises from 0.4–1.2 kcal mol−1 in amine to 2.3 kcal mol−1 in its protonated structure. Along with the ΔEdisp, these stabilizing interactions make the overall interaction energy favourable (ΔEint = −5.43 kcal mol−1) by overcoming the increase in the steric repulsion. The ΔEint is just slightly attenuated by the small positive ΔEprep which amounts 0.19 kcal mol−1. The charge transfer part of intramolecular hydrogen bond in g-CEAH (2.1 kcal mol−1) is significantly smaller than in CEH (see above), while that in other gauche forms of (protonated) amines does not exceed 0.6 kcal mol−1.
If the gauche effect in CEAH is compared to that in FEAH, it is smaller. This is mainly because of the decrease in the favourable electrostatic energy change (ΔEelstat = −9.45 kcal mol−1 for FEAH and ΔEelstat = −6.65 kcal mol−1 for CEAH), while the increase in the steric repulsion is smaller than that in FEAH (Table 9). The orbital interaction term is decreased, too, but less so than ΔEelstat, while stabilizing contribution of dispersion forces become larger by 0.8 kcal mol−1. The data in Table 10 reveal that the a-CEAH → g-CEAH isomerization benefits from stereoelectronic factors, as well, though less than the corresponding isomerization in FEAH. This should be attributed mainly to the increase in the C–Cl → C–N* interaction (5.3 kcal mol−1) in anti conformer compared to the corresponding C–F → C–N* interaction (2.4 kcal mol−1), which makes the overall stabilization energy change less favourable for chloro than for the fluoro derivative.
Comparison with NBO energy decomposition analysis
To be sure that conclusions of the work are not method-dependent, we also performed another energy decomposition analysis based on the NBO approach which treats a molecule as a single entity, that is it does not involve breaking of covalent bond. In the NBO energy decomposition analysis (NBOEDA), anti → gauche isomerization energy (ΔE) was decomposed into Lewis (ΔELewis) and non-Lewis (ΔEdeloc) energy changes (eqn (3)), by using the NOSTAR keyword.38 The Lewis energy corresponds to the energy of a hypothetical fully localized species, having all orbitals doubly occupied. The non-Lewis or delocalization energy represents all electron delocalization contributions to the conformational isomerization.| ΔE = ΔELewis + ΔEdeloc | (3) |
The Lewis energy change comprises steric, electrostatic and structural effects. It was further decomposed into the steric (exchange repulsion) energy (ΔEsteric) and electrostatic-structural energy (ΔEelstat+struct) by using the STERIC keyword in the NBO analysis.38 Thus, the overall conformational energy is partitioned into the three energy components (eqn (4) and Table S11 in the ESI†). Table S11† also contains the LMOEDA results for comparison.
| ΔE = ΔEelstat+struct + ΔEsteric + ΔEdeloc | (4) |
The NBO energy decomposition has been applied to study the rotational barrier in ethane,41 anomeric effect,42 and conformational isomerization.3c,43
The NBOEDA results in Table S11† are qualitatively in agreement with the LMOEDA analysis, and they do not change conclusions of the work. These results support the finding that steric repulsion is smaller for chlorine derivatives, which is the result of structural changes taking place during the conformational isomerization. The only exceptions to this observation are the aa-CEH → ga-CEH and ag-CEH → gg-CEH rotations, for which the LMOEDA also shows comparable results. In addition, in the NBOEDA analysis the negative ΔEelstat+struct term means that gauche conformation is favoured by the sum of electrostatic and structural energies. The data in Table S11† show that this energy term is negative in all cases except for the ag′-CEH → gg′-CEH and ag′-CEA → gg′-CEA isomerizations, which could be ascribed to smaller electrostatic stabilization already observed for chlorine atom in the LMOEDA analysis.
Conclusions
In this paper, the origin of the gauche preference in 2-fluoroethanol, 2-chloroethanol, 2-fluoroethylamine, 2-chloroethylamine and their protonated forms, as well as the origin of anti preference in 1-chloro-2-fluoroethane are addressed from the standpoint of an energy decomposition analysis. In the case of (protonated) alcohols and amines, energies of gauche conformers were evaluated with respect to their corresponding anti conformers having the same conformational arrangement around the C–O and C–N bonds (anti, + gauche or − gauche). It has been found that the main factor leading to the gauche effect in alcohols, protonated alcohols and protonated amines is the electrostatic energy ΔEelstat. Next come orbital interactions, involving pronounced charge transfer in hydrogen bonding only in the case of protonated 2-chloroethanol. In amines, the relative importance of ΔEelstat and ΔEpol depends on the type of conformational isomerization. Contribution of dispersion energy is usually the smallest one. The energies of these stabilizing interactions exceed the energy rise due to the increased Pauli repulsion in gauche forms. Structural changes occurring during the anti → gauche rotation of the heavy atom chain are energy consuming, but are overriden by the favourable interaction energy term. All gauche preferences also benefit from stereoelectronic effects, that is vicinal hyperconjugative interactions between XH2C and CH2Y fragments, except in the case of protonated 2-chloroethanol for which aa-CEH → ga-CEH and ag-CEH → gg-CEH isomerizations are not followed by an increase in the hyperconjugation.Results from this work also show that electrostatic attractive interactions in gauche forms exceed those in anti forms even when our chemical intuition tells us that gauche conformers would suffer from dipolar and lone pair repulsion. This indicates that electrostatic interactions should better be considered as an all-charge phenomenon, because such partial interactions between bond dipoles or lone pairs cannot always account for the overall electrostatic energy change following conformational isomerization. In addition, electrostatic attraction in gauche forms relative to the anti ones is more important for more electronegative and smaller elements. That is, it decreases or even reverses its sign (becomes unfavourable contribution) when fluorine is substituted with chlorine, or when oxygen is substituted with nitrogen. In fact, anti preference in CFE and smaller gauche preference in chloro derivatives do not have their origin in the intuitively anticipated larger steric repulsion. They stem from a decrease in electrostatic stabilization upon going from anti to gauche structures, as well as a decrease in the orbital interaction energy. The former is often (somewhat) more pronounced. Or, if we still want to stick to some partial interactions, our results show that bringing smaller electronegative atoms close to each other results in nucleus–electron attraction overcoming the electron–electron and nucleus–nucleus repulsion. The relative magnitude of these attractive and repulsive interactions decreases, or even become reversed with increasing size and decreasing electronegativity of interacting atoms.
Our conclusions about the influence of atom size on the strength of electrostatic attraction related to anti → gauche conformational isomerization differ somewhat from conclusions from a very detailed analysis of chemical bonding in diatomic first and second octal row elements, which emphasized that electrons in more diffuse orbitals contribute more to the net electrostatic attraction, though orbital shape also has an influence.34c
Acknowledgements
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grant no. 172020).References
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- E. L. Eliel, S. H. Wilen and L. N. Mander, in Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York, 1994, pp. 606–615 Search PubMed.
- (a) P. R. Rablen, R. W. Hoffmann, D. A. Hrovat and W. Thatcher Borden, J. Chem. Soc., Perkin Trans. 2, 1999, 1719–1726 RSC; (b) B. M. Wong, M. M. Fadri and S. Raman, J. Comput. Chem., 2008, 29, 481–487 CrossRef CAS PubMed; (c) F. R. Souza, M. P. Freitas and R. Rittner, J. Mol. Struct.: THEOCHEM, 2008, 863, 137–140 CrossRef CAS PubMed; (d) D. Nori-Shargh and J. E. Boggs, Struct. Chem., 2011, 22, 253–262 CrossRef CAS.
- S. Wolfe, Acc. Chem. Res., 1972, 5, 102–111 CrossRef CAS.
- K. B. Wiberg, M. A. Murcko, K. E. Laidig and P. J. MacDougall, J. Phys. Chem., 1990, 94, 6956–6959 CrossRef CAS.
- (a) L. Goodman, H. Gu and V. Pophristic, J. Phys. Chem. A, 2005, 109, 1223–1229 CrossRef CAS PubMed; (b) I. V. Alabugin, K. M. Gilmore and P. W. Peterson, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 109–141 CrossRef CAS.
- M. Baranac-Stojanović, RSC Adv., 2014, 4, 43834–43838 RSC.
- C. R. S. Briggs, D. O'Hagan, H. S. Rzepa and A. M. Z. Slawin, J. Fluorine Chem., 2004, 125, 19–25 CrossRef CAS PubMed.
- D. O'Hagan, C. Bilton, J. A. K. Howard, L. Knight and D. J. Tozer, J. Chem. Soc., Perkin Trans. 2, 2000, 605–607 RSC.
- D. Y. Buissonneaud, T. Van Mourik and D. O'Hagan, Tetrahedron, 2010, 66, 2196–2202 CrossRef CAS PubMed.
- C. Sparr, E. Salamanova, W. B. Schweizer, H. M. Senn and R. Gilmour, Chem.–Eur. J., 2011, 17, 8850–8857 CrossRef CAS PubMed.
- (a) J. Huang and K. Hedberg, J. Am. Chem. Soc., 1989, 111, 6909–6913 CrossRef CAS; (b) D. A. Dixon and B. E. Smart, J. Phys. Chem., 1991, 95, 1609–1612 CrossRef CAS; (c) J. M. Bakke, L. H. Bjerkeseth, T. E. C. L. Rønnow and K. Steinsvoll, J. Mol. Struct., 1994, 321, 205–214 CrossRef CAS; (d) C. Trindle, P. Crum and K. Douglass, J. Phys. Chem. A, 2003, 107, 6236–6242 CrossRef CAS; (e) C. R. S. Briggs, M. J. Allen, D. O'Hagan, D. J. Tozer, A. M. Z. Slawin, A. E. Goeta and J. A. K. Howard, Org. Biomol. Chem., 2004, 2, 732–740 RSC; (f) F. R. Souza and M. P. Freitas, Comput. Theor. Chem., 2011, 964, 155–159 CrossRef CAS PubMed; (g) P. I. Nagy, J. Phys. Chem. A, 2013, 117, 2812–2826 CrossRef CAS PubMed; (h) R. A. Cormanich, R. Rittner, M. P. Freitas and M. Bühl, Phys. Chem. Chem. Phys., 2014, 16, 19212–19217 RSC.
- (a) K.-M. Marstokk and H. Møllendal, Acta Chem. Scand., Ser. A, 1980, 34, 15–29 CrossRef PubMed; (b) J. A. S. Smith and V. F. Kalasinsky, Spectrochim. Acta, Part A, 1986, 42, 157–167 CrossRef; (c) I. Hyla-Kryspin, S. Grimme, S. Hruschka and G. Haufe, Org. Biomol. Chem., 2008, 6, 4167–4175 RSC; (d) J. R. Durig, A. Ganguly, C. Zheng, G. A. Gurigis, W. A. Herrebout, B. J. van der Veken and T. K. Gounev, J. Mol. Struct., 2010, 968, 36–47 CrossRef CAS PubMed.
- (a) J. A. K. Howard, V. J. Hoy, D. O'Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613–12622 CrossRef CAS; (b) J. D. Dunitz and R. Taylor, Chem.–Eur. J., 1997, 3, 89–98 CrossRef CAS.
- R. A. Cormanich, M. P. Freitas, C. F. Tormena and R. Rittner, RSC Adv., 2012, 2, 4169–4174 RSC.
- (a) J. P. Snyder, N. S. Chandrakumar, H. Sato and D. C. Lankin, J. Am. Chem. Soc., 2000, 122, 544–545 CrossRef CAS; (b) D. C. Lankin, G. L. Grunewald, F. A. Romero, I. Y. Oren and J. P. Snyder, Org. Lett., 2002, 4, 3557–3560 CrossRef CAS PubMed; (c) A. Sun, D. C. Lankin, K. Hardcastle and J. P. Snyder, Chem.–Eur. J., 2005, 11, 1579–1591 CrossRef CAS PubMed; (d) N. E. J. Gooseman, D. O'Hagan, A. M. Z. Slawin, A. M. Teale, D. J. Tozer and R. J. Young, Chem. Commun., 2006, 3190–3192 RSC; (e) N. E. J. Gooseman, D. O'Hagan, M. J. G. Peach, A. M. Z. Slawin, D. J. Tozer and R. J. Young, Angew. Chem., Int. Ed., 2007, 46, 5904–5908 CrossRef CAS PubMed.
- (a) L. E. Zimmer, C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 11860–11871 CrossRef CAS PubMed; (b) E.-M. Tanzer, L. E. Zimmer, W. B. Schweizer and R. Gilmour, Chem.–Eur. J., 2012, 18, 11334–11342 CrossRef CAS PubMed; (c) E.-M. Tanzer, W. B. Schweizer, M.-O. Ebert and R. Gilmour, Chem.–Eur. J., 2012, 18, 2006–2013 CrossRef CAS PubMed; (d) Y. P. Rey, L. E. Zimmer, C. Sparr, E.-M. Tanzer, W. B. Schweizer, H. M. Senn, S. Lakhdar and R. Gilmour, Eur. J. Org. Chem., 2014, 1202–1211 CrossRef CAS.
- (a) J. Huang and K. Hedberg, J. Am. Chem. Soc., 1990, 112, 2070–2075 CrossRef CAS; (b) J. R. Durig, J. Liu and T. S. Little, J. Phys. Chem., 1991, 95, 4664–4672 CrossRef CAS; (c) C. Cappelli, S. Corni and J. Tomasi, J. Phys. Chem. A, 2001, 105, 10807–10815 CrossRef CAS.
- (a) A. Almenningen, L. Fernholt and K. Kveseth, Acta Chem. Scand., Ser. A, 1977, 31, 297–305 CrossRef PubMed; (b) J. R. Durig, L. Zhou, T. K. Gounev, P. Klaeboe, G. A. Guirgis and L.-F. Wang, J. Mol. Struct., 1996, 385, 7–21 CrossRef CAS; (c) S. X. Tian, N. Kishimoto and K. Ohno, J. Phys. Chem. A, 2003, 107, 53–62 CrossRef CAS.
- H. Møllendal, S. Samdal and J.-C. Guillemin, J. Phys. Chem. A, 2011, 115, 4334–4341 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS , Gamess (2013-R1 version) was used.
- (a) K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS; (b) K. Morokuma, Acc. Chem. Res., 1977, 10, 294–300 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fronseca Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- F. M. Bickelhaupt and E. J. Baerends, Angew. Chem., Int. Ed., 2003, 42, 4183–4188 CrossRef CAS PubMed.
- A. Szabó, A. Kovács and G. Frenking, Z. Anorg. Allg. Chem., 2005, 631, 1803–1809 CrossRef.
- H. Jacobsen and T. Ziegler, J. Am. Chem. Soc., 1994, 116, 3667–3679 CrossRef CAS.
- (a) M. El-Hamdi, W. Tiznado, J. Poater and M. Solà, J. Org. Chem., 2011, 76, 8913–8921 CrossRef CAS PubMed; (b) M. Baranac-Stojanović, Chem.–Eur. J., 2014, 20, 16558–16565 CrossRef PubMed.
- J. Poater, R. Visser, M. Solà and F. M. Bickelhaupt, J. Org. Chem., 2007, 72, 1134–1142 CrossRef CAS PubMed.
- (a) D. Cappel, S. Tüllmann, A. Krapp and G. Frenking, Angew. Chem., Int. Ed., 2005, 44, 3617–3620 CrossRef CAS PubMed; (b) I. Fernández and G. Frenking, Chem.–Eur. J., 2006, 12, 3617–3629 CrossRef PubMed; (c) I. Fernández and G. Frenking, Faraday Discuss., 2007, 135, 403–421 RSC.
- (a) C. Esterhuysen and G. Frenking, Theor. Chem. Acc., 2004, 111, 381–389 CrossRef CAS PubMed; (b) A. Kovács, C. Esterhuysen and G. Frenking, Chem.–Eur. J., 2005, 11, 1813–1825 CrossRef PubMed; (c) A. Krapp, F. M. Bickelhaupt and G. Frenking, Chem.–Eur. J., 2006, 12, 9196–9216 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013 Search PubMed.
- N. C. Craig, A. Chen, K. Hwan Suh, S. Klee, G. C. Mellau, B. P. Winnewisser and M. Winnewisser, J. Am. Chem. Soc., 1997, 119, 4789–4790 CrossRef CAS.
- H. Takeo, C. Matsumura and Y. Morino, J. Chem. Phys., 1986, 84, 4205–4210 CrossRef CAS PubMed.
- For details, see: (a) E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CrossRef CAS; (b) F. Weinhold and C. R. Landis, in Discovering Chemistry with Natural Bond Orbitals, John Wiley & Sons, Inc., 2012 Search PubMed; (c) F. Weinhold, NBO Manual, Board of Regents of the University of Wisconsin System on behalf of the Theoretical Chemistry Institute, 1996–2008 Search PubMed.
- H.-W. Xi, M. Karni and Y. Apeloig, J. Phys. Chem. A, 2008, 112, 13066–13079 CrossRef CAS PubMed.
- (a) Y. Mo, W. Wu, L. Song, M. Lin, Q. Zhang and J. Gao, Angew. Chem., Int. Ed., 2004, 43, 1986–1990 CrossRef CAS PubMed; (b) Y. Mo and J. Gao, Acc. Chem. Res., 2007, 40, 113–119 CrossRef CAS PubMed.
- L. Goodman, H. Gu and V. Pophristic, J. Chem. Phys., 1999, 110, 4268–4274 CrossRef CAS PubMed.
- M. P. Freitas, Org. Biomol. Chem., 2013, 11, 2885–2890 CAS.
- (a) L. A. F. Andrade, J. M. Silla, C. J. Duarte, R. Rittner and M. P. Freitas, Org. Biomol. Chem., 2013, 11, 6766–6771 RSC; (b) K. M. S. Gonçalves, D. R. Garcia, T. C. Ramalho, J. D. Figueroa-Villar and M. P. Freitas, J. Phys. Chem. A, 2013, 117, 10980–10984 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized structural parameters, comparison of LMOEDA and NBOEDA results, absolute energies and x, y, z coordinates of the studied molecules. See DOI: 10.1039/c5ra01164g |
| This journal is © The Royal Society of Chemistry 2015 |