DOI:
10.1039/C5RA01118C
(Paper)
RSC Adv., 2015,
5, 20650-20655
Facile synthesis of meso-structured Pd/FeOx and its highly catalytic performance for low temperature CO oxidation under ambient conditions†
Received
20th January 2015
, Accepted 17th February 2015
First published on 17th February 2015
Abstract
A series of meso-structured Pd/FeOx catalysts were successfully fabricated through a facile pyrolysis and in situ reduction strategy. The as-prepared materials possessed relatively high surface area and highly dispersed Pd species, and exhibited excellent low temperature CO oxidation properties under ambient conditions. Complete CO conversion could be achieved at as low as 0 °C, when 2.5 vol% H2O was introduced into the feed gas. In situ DRIFT analysis proved that these excellent catalytic properties can be attributed to the promotion of the water molecules and the synergetic effect between Pd nanoparticles and the meso-structured FeOx support.
Introduction
Low temperature CO oxidation has been studied extensively in the past two decades due to its great importance for both practical application and fundamental research.1–6 In practical applications, especially for automotive emission controlling processes, a large amount of CO is usually generated during the cold start period, entering the air within exhaust emissions and thus causing serious environmental problems. Therefore, it is of great necessity to regulate CO emission or even eliminate the diluted CO in the polluted air by developing efficient catalysts that convert CO into CO2 under ambient condition. Although many catalytic systems for low temperature CO oxidation have been well developed in recent years, most of them are easily deactivated by the moisture.7–18 Recent studies have suggested that platinum metals have much higher water residence during CO oxidation process. Thus, much attention has been focused on the supported noble metal catalyst, especially the supported Pd(O) catalyst.19–23
It is well known that the synergism between active component and support plays an important role in determining the catalytic performance. Benefited from the various oxidation states, charge-variable transition metal oxide possesses excellent performance in oxygen storage and release which is also critical factor for low temperature CO oxidation. When combined with Pd nanoparticles, significantly enhanced activity could be achieved especially under moisture conditions. This has been well proved in our previous studies.24,25 In the mixed iron oxide, there is two different oxidation state ferrum ions, which not only benefit its electron transferring, but also possess much higher oxygen storage and release capacity. Recently, Deng et al. reported that Fe(OH)x or FeOx supported Pd catalyst were more active for low temperature CO oxidation reaction under dry condition.26 It is therefore expected that mesoporous FeOx supported Pd catalyst should give the excellent catalyst for CO oxidation under moisture condition.
In this work, meso-structured FeOx support was synthetized through the facile thermal decomposition of oxalate precursor FeC2O4·2H2O. The palladium nanoparticles were successfully and homogeneously loaded by an improved wet impregnation with an in situ reduction protocol. The as-prepared materials possessed relatively high specific surface area and the palladium nanoparticles were homogeneously dispersed in the mesoporous support. The catalytic activity of prepared Pd/FeOx materials with different Pd loading contents were measured under dry and moisture condition, respectively.
Experimental
Material preparation
10 mmol FeSO4·7H2O was first dissolved in 100 ml aqueous solution, then 10 ml 1 M oxalate was slowly added under vigorous stirring. The resulting precipitate was filtered, washed with deionized water and dried at 60 °C for 24 h. The mesporous FeOx support was obtained after calcinated at 250 °C for 2 h.
A series of FeOx supported Pd catalysts with different loading content were synthesized through an improved wet impregnation method. In a typical synthesis, 1 g mesoporous FeOx material was added in 20 ml aqueous solution containing desired content of Na2PdCl4. After stirred for 2 h, 500 ml 0.1 M hydrazine hydrate (NH2NH2·H2O) was added dropwise under vigorous stirring. The resulting precipitate was centrifuged, washed several times with deionized water and dried at 60 °C for 24 h.
Characterization
Powder X-ray diffraction was measured on a Bruker D8 Focus powder diffract meter with graphite monochromatized Cu Kα radiation (λ = 0.15405 nm) operated at 40 kV. Thermo gravimetric analysis (TG-DSC) was recorded from ambient temperature to 800 °C at a heating rate of 10 °C min−1 with an air flow rate of 20 ml min−1. Nitrogen adsorption and desorption isotherms were obtained by a Micromeritics TriStar II 3020 analyzer at 77 K. The specific surface area and pore size distribution were calculated using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. High-Resolution Transmission Electron Microscopy (HRTEM) observations were performed on a field emission JEM-2100 (JEOL) electron microscope operated at 300 kV equipped with a Gatan-666 electron energy loss spectrometer and energy dispersive X-ray spectrometer. XPS (X-ray photoelectron spectroscopy) signals were collected on a VG Micro MK II instrument using monochromatic Al Kα X-ray at 1486.6 eV operated at 200 W. All the elemental binding energies were referenced to the C (1s) line situated at 284.6 eV. H2 temperature programmed reduction (H2-TPR) analysis was performed by using a Micromeritics ChemiSorb 2750 apparatus. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) spectra were recorded by a FTIR spectrometer (Nicolet iS10) equipped with a MCT detector. The sample cell was fitted with ZnSe windows. The sample was exposed to the corresponding reactive stream (60 ml min−1). The DRIFTS spectra obtained at 25 °C with a solution of 4 cm−1 and 50 scans. Typical gas mixture was 0.5 vol% CO, 20.0 vol% O2 balanced with He. Water vapor was carried into the gas mixture by a bubbler in a water bath at room temperature. After a designated amount of time, the CO flow was switched to a humid stream containing 1.5 vol% water.
Catalytic activity test
The catalytic activity for CO oxidation was measured in fixed-bed quartz tubular reactor (i.d. = 6 mm) containing 100 mg of catalyst without any pretreatment. A mixture contained 1.0 vol% CO, 20.0 vol% O2 and high-purity N2 (99.99%) was used as source gas. The feed gas at a flow rate of 25 ml min−1 was introduced into the reactor using mass-flow controllers, corresponding to a space velocity of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ml h−1 g−1. The conversion of CO was calculated by online gas chromatograph (GC). The reaction temperature was monitored through a thermocouple installed in the catalyst bed. The catalytic activity in moisture condition was tested by passing the feed gas at a flow rate of 75 ml min−1 through a water vapor saturator.
000 ml h−1 g−1. The conversion of CO was calculated by online gas chromatograph (GC). The reaction temperature was monitored through a thermocouple installed in the catalyst bed. The catalytic activity in moisture condition was tested by passing the feed gas at a flow rate of 75 ml min−1 through a water vapor saturator.
Results and discussion
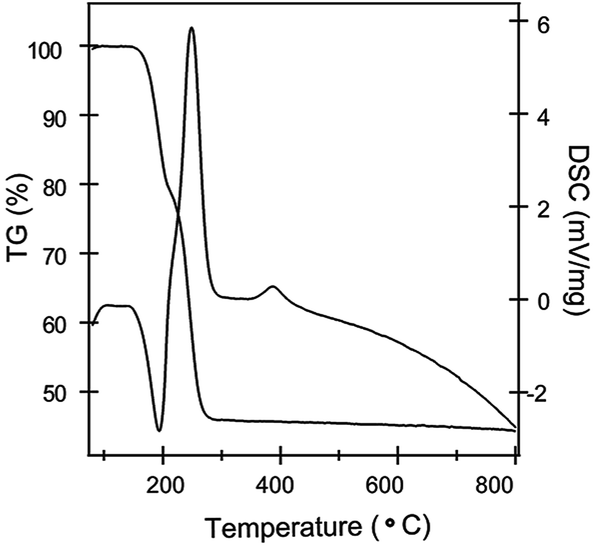
Meso-structured FeOx support was fabricated directly through controlled thermal decomposition of ferrous oxalate. The decomposition process has a strong effect on crystallite size and the texture of the product, in addition to the catalytic activities. In order to determine an appropriate temperature for calcination, the thermal behaviour of the ferrous oxalate precursor was first investigated by TG-DSC analysis in a static air atmosphere at a heating rate of 10 °C min−1, as shown in Fig. 1. From ambient temperature to 800 °C, the oxalate precursor lost 54% of its original weight in three steps accompanied by two endothermic and two exothermic process. The first two endothermic peak occurring at about 78 °C and 195 °C correspond to the elimination of absorbed and coordinated water in the precursor, respectively. Further increasing the temperature to 230 °C, there is a rapid weight loss companied with a strong exothermic peak in the DSC line, suggesting the decomposition of the oxalate into crystalline FeOx and the combustion of the decomposition product. The little exothermic peak at 400 °C in the DSC line should be contributed to the phase transformation between different Fe oxides. The temperature for calcination was determined at 250 °C in the experiment to get the meso-structured mixed phase FeOx support.
 |
| | Fig. 1 The TG-DSC curves of the FeC2O4·2H2O precursor. | |
Crystal phase composition
Fig. 2A depicts the powder X-ray diffraction (XRD) pattern of the ferrous oxalate precursor, a series of sharp and symmetrical Bragg diffraction peaks can be found in the wide angle area, which demonstrates the well crystallized feature. After calcined at 250 °C for 2 h, the ferric oxalate precursor was completely decomposed and converted to ferric oxide. (Fig. 2B). It clearly shows that the main components of the resulting oxide are hematite (Fe2O3). Besides, some weak peaks marked with triangle appearing at 2θ = 30.2°, 35.5°, 43.2°, 47.2° also indicate the existence of Fe3O4. When loaded with Pd, two peaks emerged at 2θ = 40° and 46°, which could be identified as (111) and (200) diffraction bands of face-centered cubic structure palladium, respectively. Diffraction intensity increased with the increase of Pd loading content. It should be noted that all the characteristic peaks of metallic Pd were broad and weak, indicating highly dispersed amorphous or small palladium nanoparticles. However, due to the partial overlapping between diffraction peaks of Pd (111) and Fe2O3 (004), it is difficult to estimate palladium particle size from Pd (111) using the Scherrer equation. The actual contents of Pd loaded in the meso-structured Pd/FeOx materials were determined by the ICP-AES technique to be 1.1 wt%, 3.3 wt%, 5.8 wt%, 7.1 wt%, 9.0 wt%.
 |
| | Fig. 2 The XRD patterns of FeC2O4·2H2O precursor (A) and Pd/FeOx catalysts with different Pd loading contents (B): 0 wt% (a), 1.1 wt% (b), 3.3 wt% (c), 5.8 wt% (d), 7.1 wt% (e) and 9.0 wt% (f). | |
Morphology, pore structure and surface area
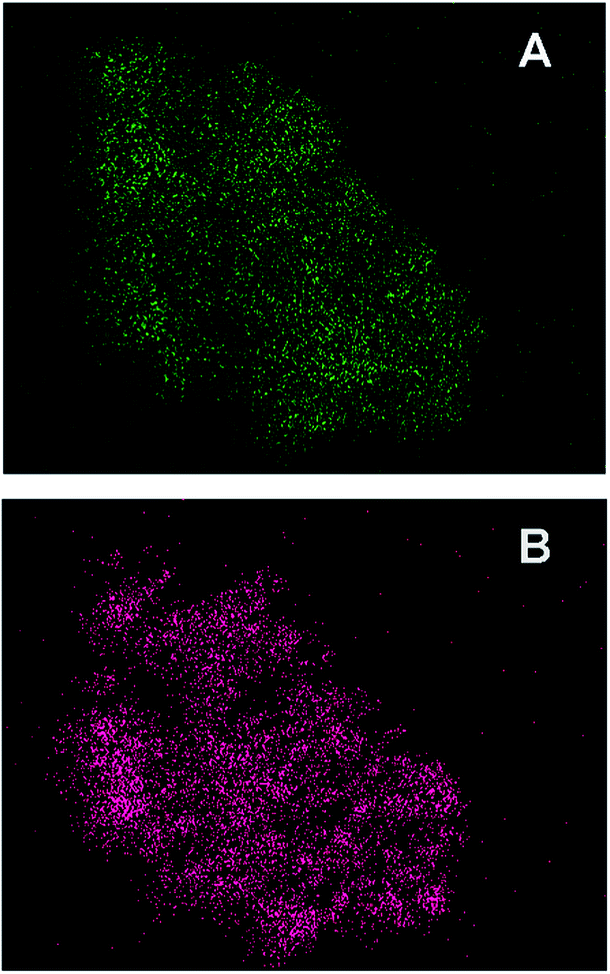
To get more explicit details, transmission electron microscopy (TEM) were carried out to observe the morphology of Pd/FeOx and the distribution of Pd in the materials (Fig. 3). For FeOx support, 10–15 nm nanoparticles uniformly aggregated together to form the meso-pore, which were generated from the decomposition and mass loss of ferrous oxalate hydrate crystals during pyrolysis. The sequential impregnation and reduction process did not affect its porous structure. When loaded with palladium, there is not any large particles in the TEM images and the noble metal nanoparticles cannot be well distinguished, suggesting that the Pd species have been highly dispersed into or on the mesoporous FeOx support. The insets are selected area electron diffraction (SAED) patterns from the same area. The well-defined diffraction rings for Fe2O3 and Fe3O4 can be found in all the samples. Instead, the relative faint and disintegrated diffraction ring for Pd indicates its very low crystallinity and/or small size and only can be found when Pd loading content higher than 3.3 wt%, which is consistent well with above XRD result. The further high resolution transmission election microscopy (HRTEM) investigation clearly shows that the catalysts are composed of crystallized FeOx and poorly crystallized palladium nanoparticles (ESI†). The element mapping images in Fig. 4 provide a further overall observation of element distribution. The two different colours represent two kinds of element (Fe, Pd) respectively. The palladium nanoparticles has been highly dispersed into/on mesoporous supports.
 |
| | Fig. 3 The TEM images of the Pd/FeOx catalysts with different Pd loading contents: 0 wt% (A), 1.1 wt% (B), 3.3 wt% (C), 5.8 wt% (D), 7.1 wt% (E) and 9.0 wt% (F). | |
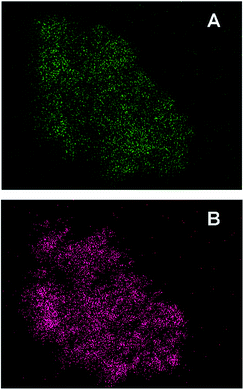 |
| | Fig. 4 The element mapping images of Pd/FeOx catalyst: Fe (A), Pd (B). | |
The texture properties of the materials were investigated by measuring adsorption and desorption isotherms of nitrogen at 77 K, as shown in Fig. 5. In all the cases, the typical Langmuir IV isotherms suggest the mesoporous structure and the appearance of H3 hysteresis loops indicates the formation of slit-like mesopores, which were generated from the decomposition and mass loss of oxalate hydrate precursor during the pyrolysis. The specific surface area of the materials was calculated by Brunauer–Emmet–Teller (BET) equation and the inset shows the pore size distribution obtained through the Barrett–Joyner–Halenda (BJH) method. The specific surface areas are all located within 60–70 m2 g−1 and the main pore sizes are all centred at about 10 nm, confirms that the loading of Pd did not change the mesoporous structure nature of FeOx support. Furthermore, it is worth noting that there is only a little limited change in the specific surface area and pore size with the increase of Pd loading amount, indicates the uniformly dispersion of palladium nanoparticles.
 |
| | Fig. 5 The N2 adsorption–desorption analysis of the FeOx support (A) and the Pd/FeOx catalysts with different Pd loading contents: 1.1 wt% (B), 3.3 wt% (C), 5.8 wt% (D), 7.1 wt% (E) and 9.0 wt% (F). | |
Surface composition, metal oxidation state
The XPS measurements were performed to detect the surface chemical states of the FeOx support and 7.1 wt% Pd loaded Pd/FeOx catalyst with and without catalytic performance. In all the cases, carbon C 1s core excitation at binding energy of 284.6 eV was taken as reference. The Fe 2p XPS spectra are shown in the Fig. 6A. There are two prominent peaks at binding energy of 711.0 eV and 724.6 eV which are recognized as Fe 2p2/3, Fe 2p1/2 respectively.27–30 Meanwhile, they both have satellite peaks: 718.5 eV for Fe 2p2/3 and 732.8 eV for Fe 2p1/2. The Fe 2p2/3 spectrum can be deconvoluted into three components at binding energy of 709.7 eV, 710.8 eV and 713.5 eV, represent Fe2+ octahedral, Fe3+ octahedral and Fe3+ tetrahedral, respectively.31–33 It clearly indicates the co-existence of Fe2+ and Fe3+ on the surface of the material. Fig. 6B shows the O 1s XPS spectra which can be resolved into two peaks: lattice oxygen (Olatt) at 529.6 eV and surface adsorbed oxygen (Oads) at 531.4 eV.34 The amount of lattice oxygen decreased as the palladium loaded which can be attributed to the more absorbed oxygen on the high dispersed palladium species. Moreover, Pd 3d XPS spectra are obtained in Fig. 6C. The typical binding energy of Pd0 and Pd2+ states are 335.1 eV, 336.7 eV, 340.4 eV and 342.1 eV, respectively.35–37 It is worth to point out that most of palladium exists as Pd0 in the Pd/FeOx catalyst. However, after catalyst test, the component of Pd2+ increased probably due to the oxidation of Pd0 in the process of catalytic performance.
 |
| | Fig. 6 The XPS spectra of the FeOx support (a) and 7.1 wt% Pd/FeOx catalyst before reaction (b), after reaction under dry condition (c) and after reaction under moisture condition (d). | |
Catalytic properties
Fig. 7 shows the reduction profiles of the catalysts. For comparison, the H2 temperature programmed reduction (H2-TPR) profile of the parent FeOx is also included. The TPR profile of FeOx is consisted of two distinguishable reduction regions. One peak at about 320 °C can be assigned to the reduction of Fe2O3 to Fe3O4. The other broad signal at 450–750 °C is the reduction of Fe3O4 to Fe0. After loaded with palladium, the reduction peak is obviously shifts to the relative lower temperature region. The significant low-temperature reduction feature strongly suggests that the FeOx substrate has been ‘activated’ to a large extent in the Pd/FeOx catalyst by Pd loading.
 |
| | Fig. 7 The H2-TPR profiles of FeOx (a) and Pd/FeOx catalysts with different Pd loading contents 1.1 wt% (B), 3.3 wt% (C), 5.8 wt% (D), 7.1 wt% (E) and 9.0 wt% (F). | |
The catalytic performances of Pd/FeOx catalysts for CO oxidation under different reaction conditions are shown in Fig. 8. The FeOx support does not show any catalytic activity with the reaction temperature lower than 100 °C under dry condition. When loaded with palladium, the catalytic activity shows a significant improvement. As Pd content reached to 7.1 wt%, the CO full conversion temperature decreased as low as 47 °C. Continuous increase the Pd content does not show better catalytic activity. The catalytic activity of Pd/FeOx catalyst is not only determined by the active component, but also the geometric structures of FeOx support and the synergy between palladium and FeOx.29–31 Generally, moisture is inevitable existence during practical application for CO elimination. It is more important to explore the catalytic performance under ambient condition. Surprisingly, the catalytic activity of Pd/FeOx does not get worse but a significantly enhancement under the moisture condition (Fig. 8B). 7.1 wt% Pd/FeOx catalyst still show the highest catalytic activity. The CO full conversion temperature can be decreased as low as 0 °C, when 2.5 vol% H2O was introduced into the feed gas. It clearly suggests the appropriate moisture is beneficial to the CO oxidation when Pd/FeOx was used as catalyst. Moreover, to gain insight into the intrinsic activities of supported Pd catalysts, TOFs normalized by the number of the surface noble atoms under differential reaction conditions were calculated. No matter under the dry or moisture condition at 0 °C, The TOFs of the 7.1 wt% Pd loaded catalyst all give the maximum values (1.41 × 10−4 s−1 under dry condition and 27.9 × 10−4 s−1 under moisture condition, respectively) among all samples synthesized.
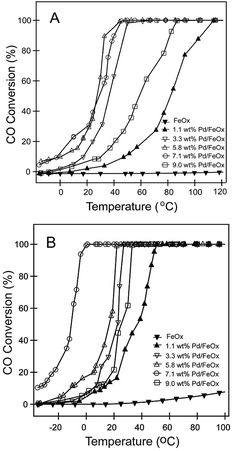 |
| | Fig. 8 The catalytic performance of FeOx and Pd/FeOx under different reaction conditions: (A) dry condition, (B) moisture condition (the water concentration in the feed gas is 2.5 vol% H2O). | |
In situ DRIFTS measurements
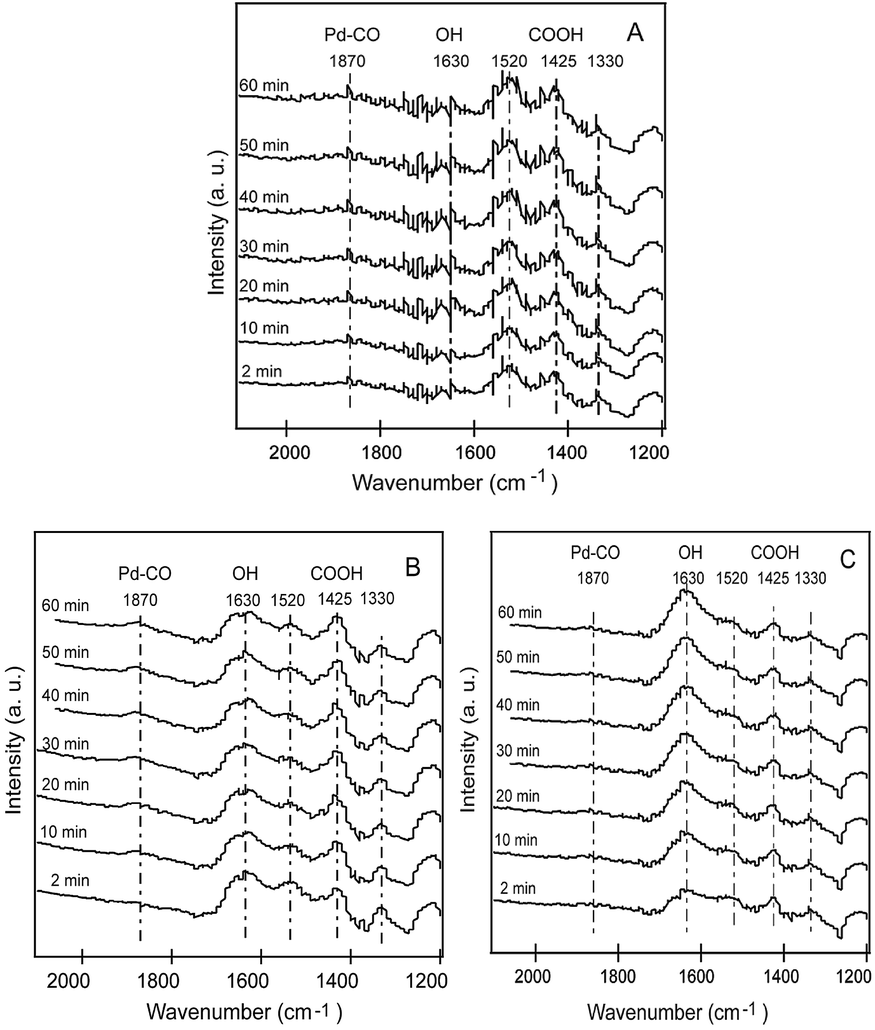
To further determine the effect of water during CO oxidation process, in situ DRIFTS technology was employed to explore the difference of surface reaction of CO on 7.1 wt% Pd/FeOx catalyst under different reaction conditions. According to the literatures, the wide bands at 1360 and 1470 cm−1 are ascribed to νs(OCO) and δ(OH) of [COOH]s, respectively.32–34 The strong band at 1600–1700 cm−1 can be assigned to the OH groups,35,36 whereas, the weak band at 1810 cm−1 is related to bonded CO on metallic Pd.37,38 Interestingly, once Pd/FeOx was exposed to 0.5 vol% CO, [COOH] species was formed immediately and the peak intensity gradually increase in the absence of water and O2, which indicates that CO directly reacts with the surface hydroxyl on Pd/FeOx catalyst to form [COOH] intermediates (Fig. 9A). When 20.0 vol% O2 was introduced into the feed gas (dry condition, Fig. 9B), the bands at 1650 cm−1 attributed to surface –OH gradually decreased with the contact time, which further prove the surface –OH group participation in the CO oxidation process. In contrast, under the moisture condition (Fig. 9C), due to the adequate supply of –OH, the intensity of –OH group remains relatively constant during the whole process. Besides, compared with that under dry stream, Pd–CO band appear and become stable more rapidly. The presence of water could not only be in favor of a formation of carbonate species (COOH), but also promote the adsorption of CO on the surface of metallic Pd. All these clearly indicate the positive effect of water in CO oxidation on the surface of Pd/FeOx catalysts.
 |
| | Fig. 9 In situ DRIFTS spectra of 7.1 wt% Pd/FeOx under (A) 0.5 vol% CO–He (B) 0.5 vol% CO–20 vol% O2–He and (C) 0.5 vol% CO–20 vol% O2–1.5 vol% H2O–He at 25 °C. | |
Conclusions
In summary, the crystalline mesoporous Pd/FeOx catalysts were successfully fabricated through an improved impregnation protocol. The as-prepared materials owned good crystallinity, relatively high specific surface area and highly dispersed Pd nanoparticles, and exhibited excellent low temperature CO oxidation properties under ambient condition. Complete CO conversion could be achieved at as low as 0 °C for 7.1 wt% Pd loaded catalyst, when 2.5 vol% H2O was introduced into the feed gas. In situ DRIFTS analysis proved the positive effect of water during CO oxidation process.
Acknowledgements
This study was supported by National Basic Research Program of China 2013CB933201.
Notes and references
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252 CrossRef CAS PubMed.
- C. T. Campbell, Science, 2004, 306, 234 CrossRef CAS PubMed.
- A. U. Nilekar, S. Alayoglu, B. Eichhorn and M. Mavrikakis, J. Am. Chem. Soc., 2010, 132, 7418 CrossRef CAS PubMed.
- A. Hadi and I. Yaacob, Catal. Today, 2004, 96, 165 CrossRef CAS PubMed.
- J. Huang, S. Wang, Y. Zhao, X. Wang, S. Wang, S. Wu, S. Zhang and W. Huang, Catal. Commun., 2006, 7, 1029 CrossRef CAS PubMed.
- M. Date, M. Okumura, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 2129 CrossRef CAS PubMed.
- R. A. Ojifinni, N. S. Froemming, J. Gong, M. Pan, T. S. Kim, J. M. White, G. Henkelman and C. B. Mullins, J. Am. Chem. Soc., 2008, 130, 6801 CrossRef CAS PubMed.
- H. H. Kung, M. C. Kung and C. K. Costello, J. Catal., 2003, 216, 425 CrossRef CAS.
- M. Date, H. Imai, S. Tsubota and M. Haruta, Catal. Today, 2007, 2, 225 Search PubMed.
- H. Yen, Y. Seo, S. Kaliaguine and F. Kleitz, Angew. Chem., Int. Ed., 2012, 51, 12032 CrossRef CAS PubMed.
- J. Zhu and Q. Gao, Microporous Mesoporous Mater., 2009, 124, 144 CrossRef CAS PubMed.
- X. Xie, L. Li, Z.-Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746 CrossRef CAS PubMed.
- Y. Feng, L. Li, S. Niu, Y. Qu, Q. Zhang, Y. Li, W. Zhao, H. Li and J. Shi, Appl. Catal., B, 2012, 111, 461 CrossRef PubMed.
- L.-N. Cai, Y. Guo, A.-H. Lu, P. Branton and W.-C. Li, J. Mol. Catal. A: Chem., 2012, 360, 35 CrossRef CAS PubMed.
- S. A. Kondrat, T. E. Davies, Z. Zu, P. Boldrin, J. K. Bartley, A. F. Carley, S. H. Taylor, M. J. Rosseinsky and G. J. Hutchings, J. Catal., 2011, 281, 279 CrossRef CAS PubMed.
- R. Xu, X. Wang, D. Wang, K. Zhou and Y. Li, J. Catal., 2006, 237, 426 CrossRef CAS PubMed.
- E. C. Njagi, C. H. Chen, H. Genuino, H. Galindo, H. Huang and S. L. Suib, Appl. Catal., B, 2010, 99, 103 CrossRef CAS PubMed.
- B. Qiao, L. Liu, J. Zhang and Y. Denga, J. Catal., 2009, 261, 241 CrossRef CAS PubMed.
- L. Liu, F. Zhou, L. Wang, X. Qi, F. Shi and Y. Deng, J. Catal., 2010, 274, 1 CrossRef CAS PubMed.
- L.-C. Wang, Q. Liu, X.-S. Huang, Y.-M. Liu, Y. Cao and K.-N. Fan, Appl. Catal., B, 2009, 88, 204 CrossRef CAS PubMed.
- A. V. Salker and R. K. Kunkalekar, Catal. Commun., 2009, 10, 1776 CrossRef CAS PubMed.
- J. S. Park, D. S. Doh and K. Y. Lee, Top. Catal., 2000, 10, 127 CrossRef CAS.
- G. Li, L. Li, Y. Yuan, J. Shi, Y. Yuan, Y. Li, W. Zhao and J. Shi, Appl. Catal., B, 2014, 158–159, 34 Search PubMed.
- G. Li, L. Li, Y. Yuan, J. Shi, Y. Yuan, Y. Li, W. Zhao and J. Shi, RSC Adv., 2014, 4, 35762 RSC.
- L. Liu, B. Qiao, Y. He, F. Zhou, B. Yang and Y. Deng, J. Catal., 2012, 294, 29 CrossRef CAS PubMed.
- X. W. Xie, L. Li, Z.-Q. Liu, M. Haruta and W. J. Shen, Nature, 2009, 458, 746 CrossRef CAS PubMed.
- M. Dat, M. Okumura, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 2129 CrossRef PubMed.
- R. V. Gulyaev, A. I. Stadnichenko, E. M. Slavinskaya, A. S. Ivanova, S. V. Koscheev and A. I. Boronin, Appl. Catal., A, 2010, 439–440, 41 Search PubMed.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439 CAS.
- J. L. Shi, Chem. Rev., 2013, 113, 2139 CrossRef CAS PubMed.
- B. B. Chen, C. Shi, M. Crocker, Y. Wang and A. M. Zhu, Appl. Catal., B, 2013, 132–133, 245 CrossRef CAS PubMed.
- Y. Denkwitz, A. Karpenko, V. Plazk, R. Leppelt, B. Schumacher and R. J. Behm, J. Catal., 2007, 246, 74 CrossRef CAS PubMed.
- S. Zhang, X. Li, B. Chen, X. Zhu, C. Shi and A. M. Zhu, ACS Catal., 2014, 4, 3481 CrossRef CAS.
- K. I. Choi and M. A. Vannice, J. Catal., 1991, 127, 465 CrossRef CAS.
- K. I. Choi and M. A. Vannice, J. Catal., 1991, 131, 36 CrossRef CAS.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, Phys. Chem. Chem. Phys., 2009, 11, 641 RSC.
- G. Rupprechter, H. Unterhalt, M. Morkel, P. Galletto, L. J. Hu and H. J. Freund, Surf. Sci., 2002, 502, 109 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: HRTEM images of the Pd/FeOx materials with different Pd content and the detailed XPS analysis result for 7.1 wt% Pd loaded Pd/FeOx catalyst. See DOI: 10.1039/c5ra01118c |
| ‡ Yuan Yuan and Gengnan Li contribute equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.