
Silica molybdic acid catalysed eco-friendly three component synthesis of functionalised tetrazole derivatives under microwave irradiation in water
Abstract
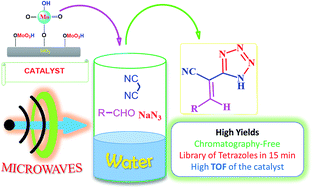
The catalytic multicomponent reaction between different aldehydes, malononitrile and sodium azide, for the synthesis of functionalised tetrazoles in pure water was performed using silica molybdic acid as an acidic catalyst under microwave irradiation at ambient temperature. The catalyst showed remarkable activity by decreasing the time period of the reaction from 24 h (without catalyst) to 15 min under microwave irradiation. The catalyst successfully tolerated different aromatic and heterocyclic aldehydes furnishing the desired products in excellent yields. The major advantages of this protocol are recyclable catalyst, water as a green solvent, excellent yields, very short reaction times, use of microwaves and high TOF of the catalyst.
 Please wait while we load your content...
Please wait while we load your content...

