
Green synthesized silver nanoparticles decorated on reduced graphene oxide for enhanced electrochemical sensing of nitrobenzene in waste water samples†
Abstract
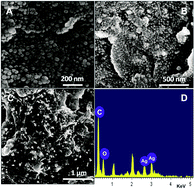
In the present work, an electrochemical sensor for nitrobenzene (NB) has been developed based on a green synthesized silver nanoparticles (AgNPs) decorated reduced graphene oxide (RGO) modified glassy carbon electrode (GCE). The AgNPs were synthesized using Justicia glauca leaf extract as a reducing and stabilizing agent. A RGO–AgNPs composite modified electrode was prepared by a simple electrochemical reduction of AgNPs dispersed GO solution. FESEM of RGO–AgNPs composite confirms that AgNPs are firmly attached on the RGO sheets and the average size of AgNPs is found to be 40 ± 5 nm. The modified electrode shows good efficiency with lower overpotential for electrocatalytic reduction of NB than that of other modified electrodes (AgNPs and RGO). The DPV response confirms that the reduction peak current of NB is linear over the concentrations from 0.5 to 900 μM. The sensitivity of the sensors is found to be 0.836 μA μM−1 cm−2 with the detection limit of 0.261 μM for NB. In addition, the RGO–AgNPs composite modified electrode shows good selectivity in the presence of potentially interfering similar compounds and good practicality in the waste water samples.
 Please wait while we load your content...
Please wait while we load your content...

