
Melt free radical grafting of glycidyl methacrylate (GMA) onto fully biodegradable poly(lactic) acid films: effect of cellulose nanocrystals and a masterbatch process
Weijun Yang,
Franco Dominici,
Elena Fortunati,
José M. Kenny and
Debora Puglia*
University of Perugia, Civil and Environmental Engineering Department, UdR INSTM, Strada di Pentima 4, 05100 Terni, Italy. E-mail: debora.puglia@unipg.it; Fax: +39 0744492950; Tel: +39 0744492916
First published on 7th April 2015
Abstract
This article reports the preparation, by means of a masterbatch procedure, of poly (lactic acid) (PLA)/cellulose nanocrystal (CNC) films via premixing 1% wt of CNC into PLA or glycidyl methacrylate (GMA) grafted PLA (g-PLA). These films were obtained by reactive extrusion and subsequent film processing. In this study, 10% wt of GMA with respect to neat PLA was used in the extrusion phase, after that a final grafting degree of 5.69% was obtained. The film obtained by using the masterbatch steps were compared with the system obtained by a direct extrusion of 1% wt of CNC in PLA/g-PLA. Thermogravimetric, crystallization and mechanical properties, as well as morphology of CNC reinforced PLA nanocomposites were characterized. Differential scanning calorimetry and thermogravimetric analysis showed enhanced crystallization ability and an improved heat resistance for the resulting nanocomposites obtained after the use of masterbatches, for example field emission scanning electron microscopy confirmed that the masterbatch preparation procedure was beneficial to the dispersion of CNC in the final nanocomposites. Furthermore, different mechanical performance was obtained when using different masterbatches, which were considered to contribute to extend the applications of PLA based composites as food packaging materials in different sectors.
Introduction
Research on biodegradable polymer nanocomposites has attracted both considerable industrial and academic attention in recent years, due to their being environmentally friendly, having good performance, design flexibility, lower life-cycle costs and a large applicability range in various industrial fields. Both the nanofillers and matrix come from bio-based, renewable agricultural resources, making them competitive with petroleum-derived materials.1–4 In the food packaging industry, there is a trend that renewable plastics will substitute petroleum based plastics with lower environmental impact, among which poly(lactic acid) (PLA) is a very promising material because of its excellent mechanical properties, transparency and commercial availability. Nonetheless, some properties, such as gas and water vapour permeability and thermal stability, are somewhat poor for some specific applications. In this context, the addition of nanoparticles as polymer additives contributes to enhance the barrier to gases due to the synergistic tortuosity, crystal nucleation and chain immobilization effects.1–5Cellulose is one of most abundant carbohydrate polymers produced by biomass, from which high modulus nanocrystals (CNC) can be extracted. These nanosized particles have been recognized as important bio-based fillers to enhance the biopolymer performance, in terms of mechanical, thermal and barrier properties. In the meanwhile, they present the advantage of being renewable when compared to other inorganic fillers, which have been widely proposed to reinforce PLA and develop biodegradable materials.1,6–9 However, homogeneous dispersion of cellulose nanoparticles is difficult to be achieved by means of traditional melt processing techniques, due to the high tendency of CNC to form agglomerates as a consequence of the presence of hydroxyl groups on the particle surfaces and their high specific surface area.10,11 In order to promote the compatibility between the components, functionalization of cellulose nanostructures and PLA can be also carried out (in solution as well as in the melt) using either surface silanization, grafting of reactive groups or polymer chains.12,13 Li et al.14 prepared a novel copolymer of PLA and glycidyl methacrylate (PLA-co-PGMA) by free radical polymerization and used it to modify the cellulose surface. Interfacial thermodynamic properties were studied and the results suggested that PLA-co-PGMA was efficient in the modification of bacterial cellulose (BC) nanofibril surface and in improvement of compatibility of PLA-cellulose composites. Fortunati et al.15 also studied the blend of PLA with an ethylene-vinyl acetate–GMA copolymer (EVA–GMA), and their composites with cellulose microfibers (CF); the results showed that the presence of EVA–GMA contributed to improve the interfacial adhesion between cellulose fibres and PLA, due to interactions of the epoxy groups of GMA with hydroxyls of CF. By using the Fourier Transform Infrared Spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM), Stenstad et al.16 confirmed that the epoxy functionality was introduced onto the microfibrillated cellulose (MFC) surface by oxidation with cerium(IV) followed by grafting of GMA and the length of the polymer chains could be varied by regulating the amount of GMA added. In a study by Martínez-Sanz et al.,17 poly(glycidyl methacrylate) (PGMA) was grafted onto bacterial cellulose nanowhiskers (BCNW) by means of a redox-initiated free radical copolymerization reaction. The neat and the PGMA-grafted BCNW were subsequently incorporated as fillers into the PLA matrix. PGMA grafting improved both matrix-filler adhesion and the dispersion of cellulose nanocrystals. The incorporation of both neat and PGMA-grafted BCNW significantly reduced the oxygen permeability of PLA. Furthermore, increased elastic modulus and tensile strength were observed for all the nanocomposites, especially when the concentration of nanocrystals was around the percolation threshold, i.e. 3% wt, but only nanocomposites containing PGMA-grafted BCNW preserved the ductility of neat PLA. Pracella et al.18 functionalized PLA and CNC by radical grafting of glycidyl methacrylate (GMA) and pre-dispersed CNC in poly(vinyl acetate) (PVAc) emulsion; their results showed that functionalized components (PLA–GMA, CNC–GMA) and/or PVAc dispersed CNC both improved the phase distribution of nanofiller and tensile properties, compared to the binary PLA/CNC nanocomposites. Most above listed researches focused on the effects of dosage and modification of nanofillers or PLA matrix on the properties of resulted nanocomposites. Jonoobi et al.,7 in order to improve the nanofiber dispersion, prepared a masterbatch of PLA/cellulose nanofibers (CNF) by solvent casting before extrusion. The morphology studies of PLA and its nanocomposites showed that a relatively good dispersion was achieved as no CNF aggregates were visible in the fracture surfaces of the PLA nanocomposites with 1 and 3% wt of CNF, hence an enhancement of the mechanical performances of the final materials was obtained. Arrieta et al.19 blended a PLA–poly(hydroxybutyrate) (PHB) masterbatch system, which was granulated into pellets and then melted in the microextruder (1 min) while 5% wt of CNC or surfactant-modified CNC (s-CNC) were subsequently added and mixed, and the results revealed that the use of a masterbatch improved the dispersion of CNC and s-CNC in the final nanocomposite films and made easier the processability between PLA and PHB.
In this study, we highlight the preparation of different masterbatches by premixing the 1% wt of CNC into PLA or modified PLA matrix and finally processed into films. Thermal and mechanical properties were tested and reported here, with the aim to evaluate their suitability for the food packaging sector, expecting to optimize the approaches to process the nanocomposites and to provide some reference for industrial manufacture and practical application of CNC reinforced bio-plastic nanocomposites.
Experimental
Materials
Poly(lactic acid) (PLA 3251D), with a specific gravity of 1.24 g cm−3, a relative viscosity of ca. 2.5, and a melt flow index (MFI) of 35 g/10 min (190 °C, 2.16 kg) was supplied by Nature Works LLC, USA. Glycidyl methacrylate (GMA 779342), with a density of 1.042 g mL−1 at 25 °C, and dicumyl peroxide (DCP 329541), with a density of 1.56 g mL−1 at 25 °C, were supplied by Sigma-Aldrich. PLA pellets were dried in an oven at 40 °C for overnight. Microcrystalline cellulose (MCC, dimensions of 10–15 μm) was supplied by Sigma-Aldrich.Methods
| MBs | Composition | Dwell time (min) |
|---|---|---|
| MB1 | g-PLA | 8 |
| MB2 | PLA + 1.17% CNC | 6 + 2 |
| MB3 | g-PLA + 6.67% CNC | 6 + 2 |
| MB4 | Neat PLA | 8 |
Grafting of GMA onto the PLA (MB1 and MB3) was performed in a twin-screw microextruder (DSM Explorer 5&15 CC Micro Compounder) in the presence of DCP as initiator. The DCP and GMA (DCP/GMA = 1/10 in weight) were mixed and the solution was sprayed onto the dried PLA matrix. The GMA content was fixed at 10% wt of the PLA weight. The mixture of GMA, PLA and initiator DCP was introduced into the microextruder. Screw speed of 100 rpm and mixing time of 8 min were used in order to realize the grafting reaction, while a temperature profile of 165–175–180 °C was chosen.
The different masterbatches were obtained by mixing various amounts of CNC with PLA or g-PLA. In details, the following processing conditions were applied:
- MB1 consisted of grafted PLA (g-PLA) prepared in the conditions above reported (total time of reaction in the extruder of t = 8 minutes);
- MB2 was obtained through incorporation of 1.17% wt of CNC into PLA matrix, with incorporation of CNC (mixing time 2 minutes) after 6 minutes of PLA heating, in order to prevent the thermal degradation for CNC;
- MB3 was obtained through incorporation of 6.67% wt of CNC into g-PLA matrix, with incorporation of CNC (mixing time 2 minutes) after 6 minutes of g-PLA heating;
- MB4 consisted of neat PLA, produced under the same parameters as described in the cases of MB1, MB2 and MB3 (total time of mixing = 8 minutes).
| Materials | MB1 (% wt) | MB2 (% wt) | MB3 (% wt) | MB4 (% wt) | CNC (% wt) | Dwell time (min) |
|---|---|---|---|---|---|---|
| PLA | 100 | 2 | ||||
| g-PLA | 100 | 2 | ||||
| PLA/1CNC | 99 | 1 | 2 | |||
| g-PLA/1CNC | 99 | 1 | 2 | |||
| g-PLA/PLA-1CNC | 15 | 85 | 2 | |||
| g-PLA-1CNC/PLA | 16 | 84 | 2 | |||
| g-PLA/PLA/1CNC | 15 | 84 | 1 | 2 |
Characterizations
| Grafted GMA (%) = VHCl × NHCl × 142.2 × 0.1/m | (1) |
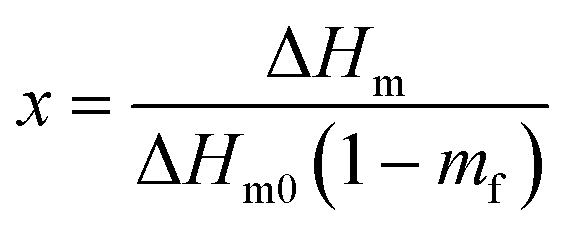 | (2) |
Results and discussion
FTIR analysis
The g-PLA (grafted polymer) was successfully prepared via free-radical polymerization, in which the peroxide initiator abstracted a tertiary hydrogen from the PLA chain to form a macromolecular radical, as shown in Fig. 1a, and the mechanism was revealed in a study by Xu,24 in which tert-butyl perbenzoate (TBPB) was used as initiator. The FTIR spectra of neat PLA, PLA/GMA, and g-PLA are given in Fig. 1b, in which PLA/GMA indicates g-PLA without further purification after the polymerization, with unreacted residual GMA inside.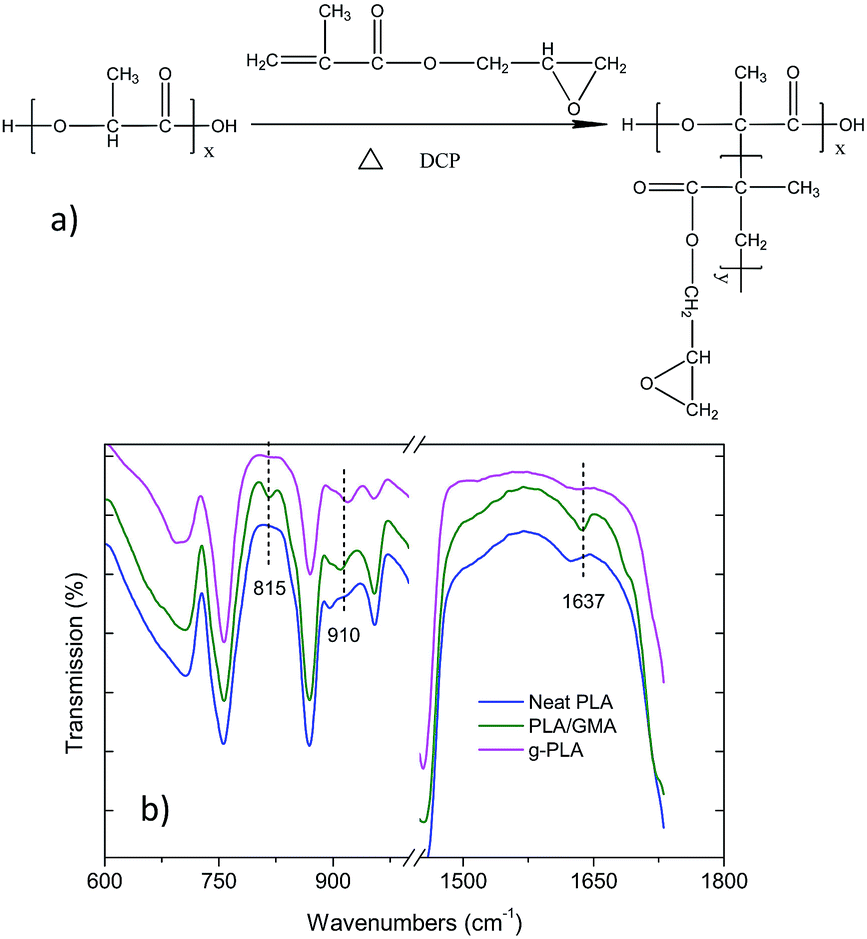 | ||
| Fig. 1 (a) Mechanism of the grafting of GMA onto the PLA chain and (b) FTIR spectra of neat PLA, PLA/GMA, and g-PLA. | ||
Compared with the spectrum of neat PLA, some new peaks appeared at 815, 910 and 1637 cm−1 in the spectrum of PLA/GMA and were associated with the carbon double bonds (815 and 1637 cm−1)24 and the asymmetric stretching of the epoxy group (910 cm−1).14,18 After eliminating the unreacted GMA, the peak of epoxy group was still present, while the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond was not observed in the case of g-PLA, which means that the CC bonds disappeared after polymerization and GMA was successfully grafted onto the PLA chain. Similar results were also reported.18
C bond was not observed in the case of g-PLA, which means that the CC bonds disappeared after polymerization and GMA was successfully grafted onto the PLA chain. Similar results were also reported.18
DSC analysis
Differential scanning calorimetry was used to investigate the glass transition (Tg), cold crystallization (Tcc), melting (Tm) and crystallinity (Xc) of PLA and PLA nanocomposites. Photos reported in Fig. 2a show the visual appearance of neat PLA and PLA nanocomposite films prepared with the different masterbatches, confirming that all the films have good transparency, with no effects related to the process used for the preparation of the different formulations. Fig. 2(b–d) and Tables 3 and 4 summarize the calorimetric parameters from the first and second heating scan for all materials.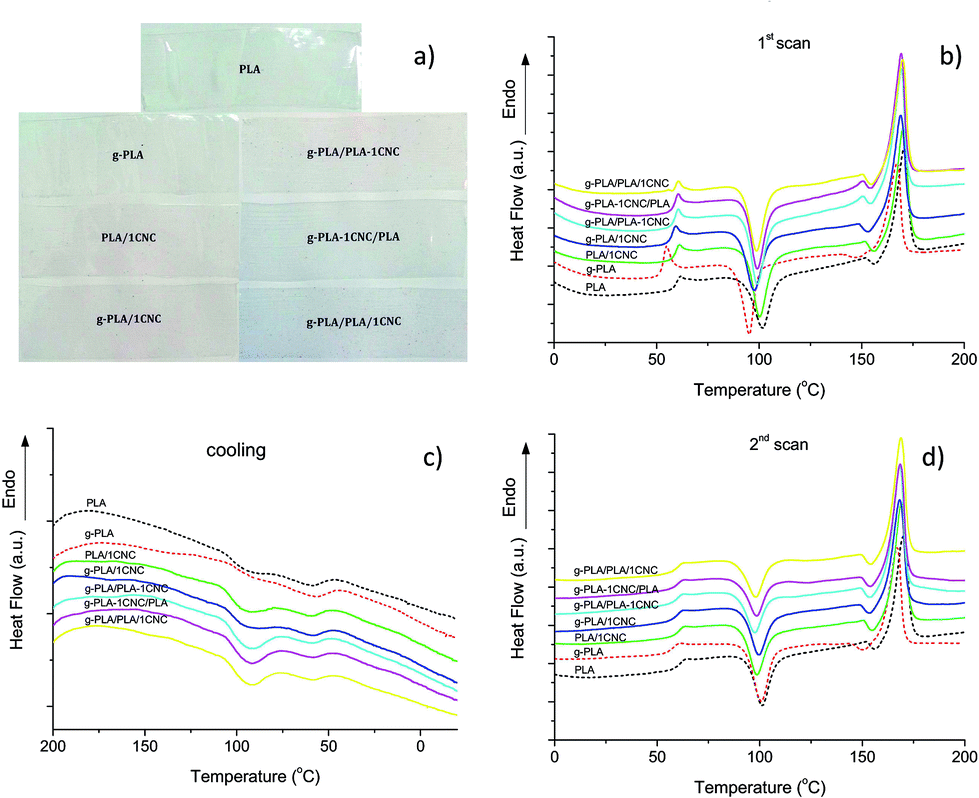 | ||
| Fig. 2 Visual appearance of films (a), DSC thermograms related to the (b) first heating scan, (c) cooling scan and (d) second heating scan of neat PLA, g-PLA and PLA nanocomposite systems obtained by different masterbatch procedures. | ||
| Materials | Tg (°C) | Tcc (°C) | Tm (°C) |
|---|---|---|---|
| PLA | 61.3 ± 0.4 | 101.8 ± 0.1 | 170.3 ± 0.2 |
| g-PLA | 54.4 ± 0.3 | 94.7 ± 0.3 | 166.9 ± 0.3 |
| PLA/1CNC | 60.9 ± 0.1 | 100.2 ± 0.1 | 170.0 ± 0.1 |
| g-PLA/1CNC | 59.4 ± 0.3 | 97.7 ± 0.5 | 168.8 ± 0.2 |
| g-PLA/PLA-1CNC | 60.6 ± 0.3 | 98.7 ± 0.3 | 169.1 ± 0.2 |
| g-PLA-1CNC/PLA | 61.0 ± 0.1 | 94.2 ± 0.1 | 169.1 ± 0.1 |
| g-PLA/PLA/1CNC | 60.5 ± 0.1 | 98.2 ± 0.4 | 169.5 ± 0.5 |
| Materials | Tg (°C) | Tcc (°C) | Tm (°C) | Xc (%) |
|---|---|---|---|---|
| PLA | 61.5 ± 0.6 | 100.3 ± 0.8 | 169.3 ± 0.2 | 15.0 ± 1.0 |
| g-PLA | 57.9 ± 0.2 | 100.1 ± 0.4 | 166.5 ± 0.5 | 7.4 ± 3.8 |
| PLA/1CNC | 60.6 ± 0.2 | 98.4 ± 0.3 | 169.0 ± 0.2 | 18.8 ± 2.6 |
| g-PLA/1CNC | 60.0 ± 0.6 | 98.9 ± 0.4 | 168.2 ± 0.1 | 16.3 ± 1.8 |
| g-PLA/PLA-1CNC | 59.2 ± 0.6 | 97.1 ± 0.7 | 168.2 ± 0.1 | 24.4 ± 0.1 |
| g-PLA-1CNC/PLA | 60.4 ± 0.3 | 97.9 ± 0.5 | 168.3 ± 0.5 | 24.1 ± 1.4 |
| g-PLA/PLA/1CNC | 59.8 ± 0.6 | 97.5 ± 0.7 | 168.4 ± 0.3 | 23.2 ± 0.7 |
To make the following discussion clearer, here we divided them into three systems, namely the first system as CNC premixed with pure PLA system (PLA, PLA/1CNC and g-PLA/PLA-1CNC), the second system as CNC premixed with g-PLA system (g-PLA, g-PLA/1CNC and g-PLA-1CNC/PLA) and the third system as the one in which CNC are introduced in the mixture without using a masterbatch procedure (g-PLA/PLA/1CNC). The low Tg, Tcc and Tm values detected for g-PLA at the first heating scan (Fig. 2b and Table 3) were due to the presence of high content of grafted PLA and residual GMA in the film after extrusion. In the case of PLA/1CNC and g-PLA/PLA-1CNC, a slight decrease of Tg was observed respect to PLA, and this effect can be related to the incorporation of CNC, that blocked the weak interactions between PLA chains, such as van der Waals force and hydrogen bond. An opposite tendency was indeed detected for g-PLA/1CNC and g-PLA-1CNC/PLA systems, in which CNC particles acted as claws. While in the case of neat g-PLA we observed a reduction in glass transition with respect of neat PLA (due to the plasticizing effect of GMA), in the case of systems that contains CNC, the presence of GMA contributed to improve the interfacial adhesion between CNC and PLA matrix, and this behavior can be explained considering the possible interactions of the epoxy groups of GMA with hydroxyls of CNC, that restrict the motion of PLA chain segments and increase the complex viscosity, with consequent increase of Tg.15 Moreover, the thermogram of g-PLA, revealed a sharper endothermic peak, respect to other samples, associated to the glass transition, which it is typically attributed to a stronger stress relaxation on heating.25 Some other studies also reported similar relaxation phenomena in plasticized PLA, confirming the presence of a compatibilizer with a plasticizing effect in the films.1,26 It was also observed that the ability to re-crystallize increased for PLA nanocomposites, with a reduction in Tcc (98.7 °C of g-PLA/PLA-1CNC and 94.2 °C of g-PLA-1CNC/PLA, respectively) in comparison with neat PLA or g-PLA, since CNC served as a preferable heterogeneous nucleation agent in PLA matrix. Moreover, the grafting of GMA onto PLA matrix promoted the dispersion of CNC in matrix, representing an important factor on the different crystallization behavior of the resulted nanocomposites.27,28
It should be also observed that the shapes of melting peaks changed at the first heating scan. In Fig. 2b, the double melting peaks appeared on the curves of g-PLA/PLA-1CNC, g-PLA-1CNC/PLA and g-PLA/PLA/1CNC, which could be attributed to the melt/recrystallization of the α form crystal of PLA.29 With the CNC premixing with PLA (MB2) and g-PLA (MB3), the melting peak at lower temperature becomes visible and the melting peak at higher temperature still retains. A small exothermic peak appeared prior to the major melting peak when the CNC were incorporated into PLA or g-PLA, due to the transition of metastable α′ form crystal to the stable α form crystal of PLA.30 Similar results were also observed with gradual increase of microfibrillated cellulose (MFC) loading in a study by Song et al.31 In that specific work, it was observed that the small exothermic peaks disappeared on the second heating scan curves. In the cooling scans shown in Fig. 2c, exothermic peaks with low intensity were observed for the pure PLA and g-PLA samples, indicating a rather low crystallization capability. In the case of PLA samples filled with 1% wt of CNC, the crystallization peaks had relatively higher intensity and started from higher temperature as a result of enhanced tendency to crystallize. The nucleation effect was remarkably enhanced when homogeneous cellulose nanocrystal dispersion in PLA and their good interaction with the matrix are achieved.32 In the second heating process (Fig. 2d and Table 4), we observed that the Tg, Tcc and Tm showed the same tendency as in the first heating scan. Specifically, it has to be observed that, even at the second heating, the combination of g-PLA with CNC favoured the nucleation effect and the crystal growth, with higher crystallinity values obtained for g-PLA/PLA-1CNC (24.4%), g-PLA-1CNC/PLA (24.1%) and g-PLA/PLA/1CNC (23.2%). DSC test results clearly evidenced that the use of a masterbatch approach, i.e. the premixing the CNC fillers with PLA or g-PLA, can be considered as an effective method to improve the CNC homogeneous dispersion and crystallization ability of resulted PLA nanocomposites.
Thermogravimetric analysis
TG and DTG curves of neat PLA and PLA based nanocomposites are shown in Fig. 3a and b, respectively. Thermal parameters including Tonset, Tmax and residual mass calculated at 600 °C were summarize in Table 5. Tonset of neat PLA was 259.0 °C, while were 262.6 and 269.2 °C for PLA/1CNC and g-PLA/PLA-1CNC, respectively. Similar tendency was also observed in the case of g-PLA, g-PLA/1CNC and g-PLA/PLA-1CNC films, indicating that both CNC and GMA improved the heat resistance of resulted nanocomposites. For g-PLA/PLA-1CNC, g-PLA-1CNC/PLA, and g-PLA/PLA/1CNC, Tonset kept the same level, however Tmax of g-PLA/PLA-1CNC (341.1 °C) and g-PLA-1CNC/PLA (348.9 °C) showed a remarkable improvement with respect of g-PLA/PLA/1CNC (330.8 °C), demonstrating that the preparation of masterbatches via pre-incorporation of 1% wt of CNC into PLA or g-PLA was a favourable method to enhance the thermal stability of CNC reinforced PLA nanocomposites. The increased Tmax for g-PLA/PLA-1CNC and g-PLA-1CNC/PLA, compared with PLA/1CNC and g-PLA/1CNC, respectively, may be attributed to the increased crystallinity due to the more homogeneous dispersion of CNC (discussed in DSC section), and this result was in agreement with the study by Shi et al.33 The reduction of Tmax for 1% wt of CNC containing PLA nanocomposites, compared with neat PLA and g-PLA, was also observed in some other studies.34–36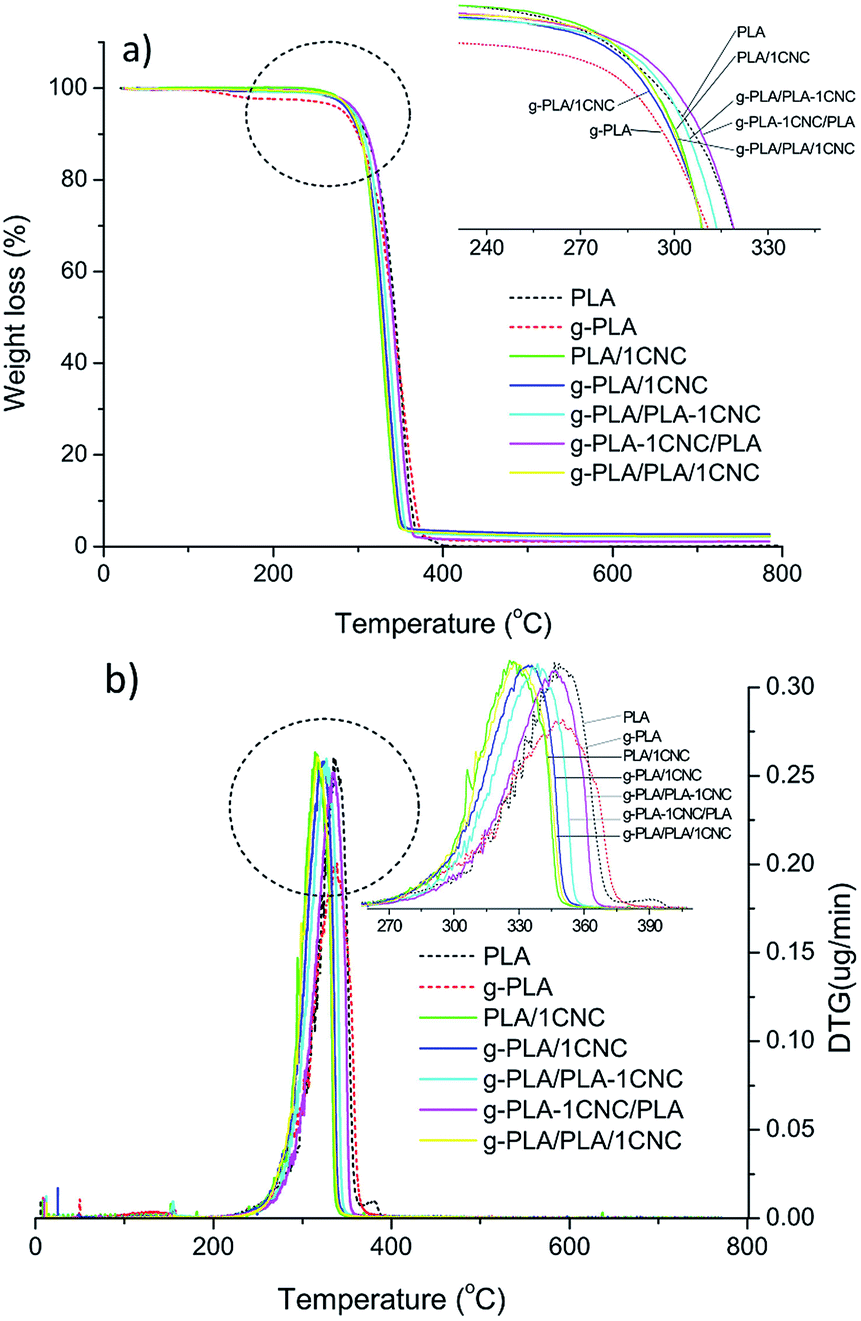 | ||
| Fig. 3 TG (a) and DTG (b) curves of neat PLA, g-PLA and PLA nanocomposite systems obtained by different masterbatch procedures. | ||
| Materials | Tonset(1%) (°C) | Tmax (°C) | Residual mass (%) @ 600 °C |
|---|---|---|---|
| PLA | 259.0 | 351.1 | 0.04 |
| g-PLA | 263.4 | 352.8 | 1.05 |
| PLA/1CNC | 262.6 | 328.2 | 2.37 |
| g-PLA/1CNC | 263.4 | 337.0 | 2.75 |
| g-PLA/PLA-1CNC | 269.2 | 341.1 | 2.21 |
| g-PLA-1CNC/PLA | 265.9 | 348.9 | 1.11 |
| g-PLA/PLA/1CNC | 268.7 | 330.8 | 2.39 |
Moreover, the addition of CNC increased the char yield (residual mass): increased char formation can limit the production of combustible gases, decreasing the exothermicity of the pyrolysis reaction, and inhibiting the thermal conductivity of the burning materials.37,38
Tensile properties
Fig. 4a–c show the tensile test results and the typical stress–strain curves (Fig. 4d) for all studied materials. The neat polymeric matrices (neat PLA and g-PLA) exhibited a step-by-step failure process, while the nanocomposites showed a brittle failure process, as a result of the incorporation of rigid CNC particles. In premixing of CNC with PLA system, the addition of 1% wt of CNC (PLA/1CNC) resulted in σ reaching 52.8 MPa, a 19% increase over the neat PLA (44.3 MPa). Furthermore, higher σ (54.2 MPa) and E (2319.0 MPa) were obtained in the case of g-PLA/PLA-1CNC, with an increase of 22.3% and 18.6% compared to PLA, respectively, due to the presence of g-PLA and better dispersion of CNC nanofillers. The same tendency could also be seen in the case of g-PLA, g-PLA/1CNC and g-PLA-1CNC/PLA, being σ and E of the two CNC containing systems increase of 26.2% and 23.7% compared with g-PLA, respectively. Obviously, the effect of changes on εb was just the opposite of that on σ and E, and it can be attributed to the presence of rigid nanocrystals, as confirmed in many previous studies.6,18,32,39 The important improvements observed in the CNC-filled PLA systems may be ascribed to the uniform distribution of CNC in the PLA matrix, and to the strong interfacial adhesion between CNC and PLA matrix. It was believed that the presence of g-PLA would inhibit self-aggregation promoting the CNC dispersion.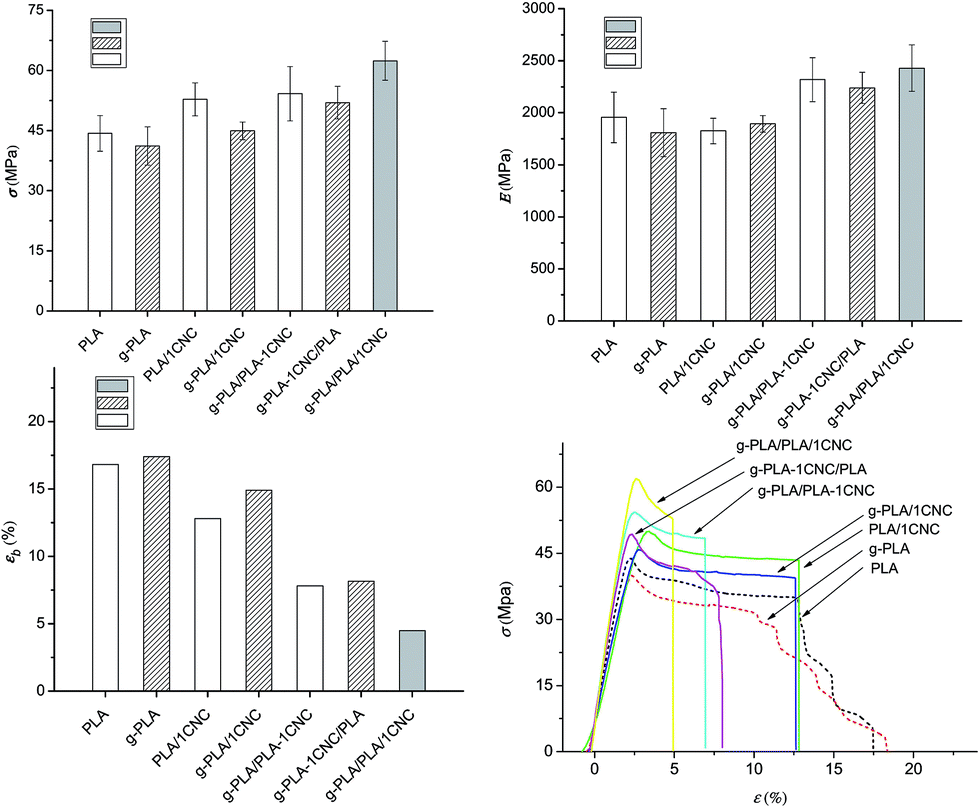 | ||
| Fig. 4 Values for tensile strength (a), Young's modulus (b), elongation at break (c) and stress–strain curves for neat PLA, g-PLA and PLA nanocomposite systems (d) obtained by different masterbatch procedures. | ||
In the g-PLA/PLA/1CNC system, maximum values of tensile strength (σ) (62.4 MPa) and elastic modulus (E) (2428.2 MPa), with a minimum of elongation at break (εb) (5.0%) were observed, confirming the creation of a rigid network formed among the cellulose nanocrystals in the nanocomposites, which greatly facilitated the enhancement of strength and modulus; at the same time, the appearance of the rigid network and self-aggregation may have slightly damaged the original PLA polymer structure, resulting in a decreased elongation of the composite.8 The tensile results were in agreement with the DSC tests, confirming how the use of masterbatches could effectively improve the mechanical performance of resulted CNC reinforced PLA nanocomposites. The different mechanical performance detected from g-PLA/PLA-1CNC, g-PLA-1CNC/PLA and g-PLA/PLA/1CNC were considered to contribute to extend applications of PLA based composites as food packaging materials in different sectors.
Morphological analysis
FESEM micrographs of CNC and fractured surfaces of the neat PLA and its nanocomposites are presented in Fig. 5. CNC aggregates (individualized with typical dimensions ranging from 100 to 200 nm in length and 5–10 nm in width) were not visible in all the fractured surfaces, indicating a good dispersion and homogeneous distribution of CNC in the nanocomposites. | ||
| Fig. 5 FESEM images of cellulose nanocrystals (CNC) and fractured samples of neat PLA, g-PLA and PLA nanocomposite systems obtained by different masterbatch procedures. | ||
While a smooth fracture surface could be seen in neat PLA, an irregular surface was observed in the PLA/1CNC system. Interestingly, the g-PLA/PLA-1CNC film showed a more flat surface compared to PLA/1CNC due to the presence of g-PLA. The fracture surface of PLA and g-PLA samples exhibited a distinct difference because of a plasticizing effect due to the presence of GMA. However, with the loading of 1% wt of CNC, a uniform surface was observed in the case of g-PLA/1CNC, suggesting a good interface adhesion between g-PLA and CNC particles. Compared to pure PLA and g-PLA, the surface of the g-PLA/PLA-1CNC and g-PLA-1CNC/PLA nanocomposites showed no significant differences, as the observed surface regularity of them was between g-PLA and g-PLA/1CNC. The PLA/PLA/1CNC system, with respect of g-PLA/PLA-1CNC and g-PLA-1CNC/PLA, had a rougher fracture surface, since some sawtooth-shape traces and cracks could be observed, which evidenced the more brittle tendency of this system to break. The observed morphological results were consistent with tensile tests (Fig. 4), confirming that the use of a masterbatch approach would be beneficial to the dispersion of cellulose nanostructures in the nanocomposites.
Conclusions
Poly(lactic acid)/cellulose nanocrystal based nanocomposites were produced by means of different masterbatch approaches, i.e. materials were prepared by extrusion of premixed 1% wt CNC in PLA or glycidyl methacrylate grafted PLA (g-PLA), followed by film processing of the different obtained formulations. The morphology studies showed that a relatively good dispersion was achieved, since smoother surfaces were visible in the fractured surfaces of the g-PLA/PLA-1CNC and g-PLA-1CNC/PLA nanocomposites when compared to g-PLA/PLA/1CNC, indicating that the grafting of GMA onto PLA matrix and the use of masterbatches were favourable methods to obtain more homogeneous distribution of CNC in PLA matrix. It was also found that the use of masterbatches was an effective approach to improve the crystallization ability of resulted PLA nanocomposites, with increased crystallinity values observed in DSC tests. The increase of temperature related to the maximum degradation rate (peaks in DTG curves) suggested that the thermal stability could be also improved applying the masterbatches procedure. The evaluation of the mechanical properties of the neat PLA and its nanocomposites showed that the tensile strength and modulus were notably improved due to the presence of g-PLA and better dispersion of CNC fillers, as an increase of 22.3% of tensile strength and 18.6% of modulus for g-PLA/PLA-1CNC was obtained in comparison with neat PLA, while values of 26.2% and 23.7% were achieved in g-PLA-1CNC/PLA composite with respect of g-PLA. Furthermore, the highest tensile strength and modulus values were registered in g-PLA/PLA/1CNC nanocomposites. This study shows that the melt compounding process using the masterbatches, in combination with the grafting of the polymeric matrix, is a very promising method to reach good processability of cellulose nanocrystal nanocomposites and achievement of improved mechanical and thermal properties for PLA. The results are supposed to contribute to extend the processing methods and applications for CNC based PLA nanocomposites.Acknowledgements
W. Y. appreciate the funding support of China Scholarship Council (CSC).Notes and references
- E. Fortunati, S. Rinaldi, M. Peltzer, N. Bloise, L. Visai, I. Armentano, A. Jiménez, L. Latterini and J. M. Kenny, Carbohydr. Polym., 2014, 101, 1122–1133 CrossRef CAS PubMed.
- N. Bitinis, R. Verdejo, J. Bras, E. Fortunati, J. M. Kenny, L. Torre and M. A. López-Manchado, Carbohydr. Polym., 2013, 96, 611–620 CrossRef CAS PubMed.
- M. Darder, P. Aranda and E. Ruiz-Hitzky, Adv. Mater., 2007, 19, 1309–1319 CrossRef CAS.
- P. Bordes, E. Pollet and L. Avérous, Prog. Polym. Sci., 2009, 34, 125–155 CrossRef CAS PubMed.
- V. P. Martino, R. A. Ruseckaite, A. Jiménez and L. Averous, Macromol. Mater. Eng., 2010, 295, 551–558 CrossRef CAS.
- N. Bitinis, E. Fortunati, R. Verdejo, J. Bras, J. M. Kenny, L. Torre and M. A. López-Manchado, Carbohydr. Polym., 2013, 96, 621–627 CrossRef CAS PubMed.
- M. Jonoobi, J. Harun, A. P. Mathew and K. Oksman, Compos. Sci. Technol., 2010, 70, 1742–1747 CrossRef CAS PubMed.
- N. Lin, J. Huang, P. R. Chang, J. Feng and J. Yu, Carbohydr. Polym., 2011, 83, 1834–1842 CrossRef CAS PubMed.
- A. Iwatake, M. Nogi and H. Yano, Compos. Sci. Technol., 2008, 68, 2103–2106 CrossRef CAS PubMed.
- J. R. Capadona, O. Van Den Berg, L. A. Capadona, M. Schroeter, S. J. Rowan, D. J. Tyler and C. Weder, Nat. Nanotechnol., 2007, 2, 765–769 CrossRef CAS PubMed.
- J. R. Capadona, K. Shanmuganathan, S. Trittschuh, S. Seidel, S. J. Rowan and C. Weder, Biomacromolecules, 2009, 10, 712–716 CrossRef CAS PubMed.
- J.-M. Raquez, Y. Murena, A.-L. Goffin, Y. Habibi, B. Ruelle, F. DeBuyl and P. Dubois, Compos. Sci. Technol., 2012, 72, 544–549 CrossRef CAS PubMed.
- K. Littunen, U. Hippi, L.-S. Johansson, M. Österberg, T. Tammelin, J. Laine and J. Seppälä, Carbohydr. Polym., 2011, 84, 1039–1047 CrossRef CAS PubMed.
- Z. Q. Li, X. D. Zhou and C. H. Pei, Int. J. Polym. Mater., 2010, 59, 725–737 CrossRef CAS.
- E. Fortunati, D. Puglia, J. M. Kenny, M. Minhaz-Ul Haque and M. Pracella, Polym. Degrad. Stab., 2013, 98(12), 2742–2751 CrossRef CAS PubMed.
- P. Stenstad, M. Andresen, B. S. Tanem and P. Stenius, Cellulose, 2008, 15, 35–45 CrossRef CAS.
- M. Martinez-Sanz, M. A. Abdelwahab, A. Lopez-Rubio, J. M. Lagaron, E. Chiellini, T. G. Williams, D. F. Wood, W. J. Orts and S. H. Imam, Eur. Polym. J., 2013, 49, 2062–2072 CrossRef CAS PubMed.
- M. Pracella, M. M.-U. Haque and D. Puglia, Polymer, 2014, 55, 3720–3728 CrossRef CAS PubMed.
- M. Arrieta, E. Fortunati, F. Dominici, E. Rayón, J. López and J. Kenny, Carbohydr. Polym., 2014, 107, 16–24 CrossRef CAS PubMed.
- E. D. Cranston and D. G. Gray, Biomacromolecules, 2006, 7, 2522–2530 CrossRef CAS PubMed.
- E. Fortunati, I. Armentano, Q. Zhou, A. Iannoni, E. Saino, L. Visai, L. A. Berglund and J. M. Kenny, Carbohydr. Polym., 2012, 87, 1596–1605 CrossRef CAS PubMed.
- J. Liu, H. Jiang and L. Chen, J. Polym. Environ., 2012, 20, 810–816 CrossRef CAS PubMed.
- O. Martin and L. Avérous, Polymer, 2001, 42, 6209–6219 CrossRef CAS.
- T. Xu, Z. Tang and J. Zhu, J. Appl. Polym. Sci., 2012, 125, E622–E627 CrossRef CAS.
- E. Fortunati, I. Armentano, A. Iannoni and J. M. Kenny, Polym. Degrad. Stab., 2010, 95, 2200–2206 CrossRef CAS PubMed.
- N. Burgos, V. P. Martino and A. Jiménez, Polym. Degrad. Stab., 2013, 98, 651–658 CrossRef CAS PubMed.
- D. Wu, L. Wu, L. Wu, B. Xu, Y. Zhang and M. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 1100–1113 CrossRef CAS.
- E. Fortunati, I. Armentano, Q. Zhou, D. Puglia, A. Terenzi, L. A. Berglund and J. M. Kenny, Polym. Degrad. Stab., 2012, 97, 2027–2036 CrossRef CAS PubMed.
- T. Dobreva, J. M. Perena, E. Pérez, R. Benavente and M. García, Polym. Compos., 2010, 31, 974–984 CrossRef CAS.
- P. Pan, B. Zhu, W. Kai, T. Dong and Y. Inoue, Macromolecules, 2008, 41, 4296–4304 CrossRef CAS.
- Y. Song, K. Tashiro, D. Xu, J. Liu and Y. Bin, Polymer, 2013, 54, 3417–3425 CrossRef CAS PubMed.
- A. Pei, Q. Zhou and L. A. Berglund, Compos. Sci. Technol., 2010, 70, 815–821 CrossRef CAS PubMed.
- Q. Shi, C. Zhou, Y. Yue, W. Guo, Y. Wu and Q. Wu, Carbohydr. Polym., 2012, 90, 301–308 CrossRef CAS PubMed.
- E. Fortunati, M. Peltzer, I. Armentano, L. Torre, A. Jiménez and J. M. Kenny, Carbohydr. Polym., 2012, 90, 948–956 CrossRef CAS PubMed.
- M. Kowalczyk, E. Piorkowska, P. Kulpinski and M. Pracella, Composites, Part A, 2011, 42, 1509–1514 CrossRef PubMed.
- E. Fortunati, F. Luzi, D. Puglia, F. Dominici, C. Santulli, J. Kenny and L. Torre, Eur. Polym. J., 2014, 56, 77–91 CrossRef CAS PubMed.
- D. Van Krevelen, Polymer, 1975, 16, 615–620 CrossRef CAS.
- R. Liepins and E. Pearce, Environ. Health Perspect., 1976, 17, 55 CrossRef CAS.
- M. Bulota, A.-H. Vesterinen, M. Hughes and J. Seppälä, Polym. Compos., 2013, 34, 173–179 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |