DOI:
10.1039/C5RA00887E
(Paper)
RSC Adv., 2015,
5, 25115-25124
Nano γ-Fe2O3-supported fluoroboric acid: a novel magnetically recyclable catalyst for the synthesis of 12-substituted-benzo[h][1,3]dioxolo[4,5-b]-acridine-10,11-diones as potent antitumor agents†
Received
15th January 2015
, Accepted 25th February 2015
First published on 26th February 2015
Abstract
Nano γ-Fe2O3-supported fluoroboric acid was synthesized as a novel magnetic catalyst and characterized. The nanoparticle reagent was obtained with good loading levels and has been successfully used for the efficient and selective synthesis of 12-substituted-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-diones. The catalyst is quantitatively recovered with an external magnet and can be reused for six cycles with almost consistent activity. In addition, all the synthetic derivatives were fully characterized using spectral data and evaluated for their antitumor activity on human hepatoma cells (HepG2) and Henrietta Lacks strain of cancer cells (HeLa). Among the 19 new compounds screened, 12-(2-fluorophenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4d) has pronounced activity.
Introduction
Along with living habits and environmental changes, cancer has become the major cause of death in both developing and developed countries. Despite the efforts to discover and develop small molecule anticancer drugs in the last decade,1 the development of new antitumor agents with improved tumor selectivity, efficiency, and safety remains desirable. Recently, naphthoquinones have been reported to posses diverse biological and therapeutic properties including antioxidant,2 antifungal,3 anti-inflammatory,4 antiviral,5 and anticancer activities.6 Among these active compounds, 1,2-naphthoquinones, including Tanshinone IIA,7 4-hydroxysaprothoquinone,8 β-lapachone,9 mansonones10 and salvicine11 (Fig. 1), have been reported to show remarkable antitumor activities by means of inhibiting multiple enzymes. One of these quinones, salvicine, is a novel diterpenoid quinone compound, which possesses potent in vitro and in vivo activities against malignant tumor cells, especially in some human solid tumor models, and has now entered phase II clinical trials.12 Salvicine induces apoptosis in various human tumor cell lines and displays prominent activity against multiple-drug resistance.13 Mechanistic studies have shown that topo II is one of the primary molecular targets of salvicine.14 These recent examples highlight the ongoing interest in new 1,2-naphthoquinone derivatives and have prompted us to investigate this pharmacophore in drug discovery programs aimed at synthesizing novel bioactive molecules.
 |
| | Fig. 1 Structures of potent anticancer 1,2-naphthoquinone analogues. | |
In recent years, magnetite or maghemite nanoparticles have drawn the attention of organic chemists as a new alternative to porous materials for supporting catalytic transformations.15 The magnetic nature of these particles allows for easy recovery and recycling of the catalysts using an external magnetic field, which may reduce operational cost and enhance product purity. Moreover, magnetic nanoparticles (MNPs) can be easily functionalized through appropriate surface modifications, which are able to load various functionalities.16
Multicomponent domino reactions have become an increasingly useful tool for the synthesis of chemically and biologically important benzoacridinediones because of their convergence, atom economy, and other suitable characteristics from the point of view of green chemistry.17 As a continuation of our interest in developing efficient and environmentally benign synthetic methodologies,18 we report herein on the preparation of a new type of magnetically separable nano γ-Fe2O3-supported fluoroboric acid (γ-Fe2O3-HBF4) and its application to the synthesis of 12-substituted-benzo[h][1,3]dioxolo[4,5-b]-acridine-10,11-diones (Scheme 1) as potent antitumor agents.
 |
| | Scheme 1 Synthesis of 12-substituted-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-diones catalyzed by γ-Fe2O3-HBF4. | |
Results and discussion
To prepare γ-Fe2O3-HBF4, we have first chosen a known nano-magnet γ-Fe2O3, which can be easily prepared by coprecipitation of ferrous (Fe2+) and ferric (Fe3+) ions in a basic aqueous solution followed by thermal treatment according to the reported procedure. Then, HBF4 was added to a suspension of γ-Fe2O3 in Et2O, with dispersion by sonication. The mixture was concentrated and the residue was heated at 100 °C for 72 h under vacuum to obtain nano γ-Fe2O3-supported fluoroboric acid (Scheme 2).
 |
| | Scheme 2 Synthesis of γ-Fe2O3-HBF4. | |
The γ-Fe2O3-HBF4 was characterized using FT-IR spectroscopy, energy dispersive spectroscopy (EDS), scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray powder diffraction (XRD), a vibrating sample magnetometer (VSM), thermogravimetric analysis (TGA) and elemental analysis. The specific surface area of the powders was determined by use of the BET method. Unfortunately, due to the magnetic properties of γ-Fe2O3-HBF4 it is actually impossible to further characterize this material using solid state NMR spectroscopy.
Fig. 2 shows the FT-IR spectra for the γ-Fe2O3 nanoparticles and γ-Fe2O3-HBF4. The FT-IR spectrum of nano-γ-Fe2O3 exhibits two characteristic peaks at 562 cm−1 and 638 cm−1 due to the stretching vibrations of the Fe–O bonds in γ-Fe2O3. The FT-IR spectrum of γ-Fe2O3-HBF4 shows Fe–O vibrations in the same vicinity. Compared with the unfunctionalized γ-Fe2O3, the significant feature observed for γ-Fe2O3-HBF4 was the appearance of peaks at 1033 cm−1 (B–F stretching vibrations). This analysis, in combination with microanalysis data (Fig. 3), indicated the successful loading of the HBF4 groups onto the magnetic nanoparticles. The amount of HBF4 loaded on the surface of nano γ-Fe2O3 was determined using TG analysis and confirmed by ion-exchange pH analysis.
 |
| | Fig. 2 FT-IR spectra of γ-Fe2O3 and γ-Fe2O3-HBF4. | |
 |
| | Fig. 3 EDS of γ-Fe2O3-HBF4. | |
The XRD pattern of γ-Fe2O3-HBF4 shows characteristic peaks and relative intensity, which match well with the cubic structure of maghemite (JCPDS file no. 39-1364). Diffraction peaks at around 30.32, 35.60, 43.30, 54.42, 57.20 and 62.98 corresponding to the (220), (311), (400), (422), (511) and (440) faces are readily recognized from the XRD pattern (Fig. 4). The average crystallite size was calculated to be 13.4 nm using the Scherrer equation. The XRD combined with elemental analysis (B 0%, F 0%) of γ-Fe2O3-HBF4 heated to 600 °C shows that all F is lost as HF/HBF4 into the gas phase and Fe fluoride is not formed.
 |
| | Fig. 4 (a) XRD of γ-Fe2O3. (b) XRD of γ-Fe2O3-HBF4. (c) XRD of γ-Fe2O3-HBF4 heated to 600 °C. | |
One indication of bond formation between the nanoparticles and the catalyst can be inferred from the TGA. TGA was also used to determine the percentage of HBF4 groups physisorbed onto the surface of the magnetic nanoparticles. The TGA curve of γ-Fe2O3-HBF4 shows the mass loss of the HBF4 group as it decomposes upon heating (Fig. 5). The weight loss at temperatures below 100 °C is due to the removal of physically adsorbed water. HBF4 groups have been reported to desorb at temperatures above 160 °C. A weight loss of about 6.6% occurs from 160–500 °C, resulting from the removal of OH and HBF4 physisorbed onto the MNPs surface.
 |
| | Fig. 5 TG analysis for γ-Fe2O3 and γ-Fe2O3-HBF4. | |
The shape and surface morphology of γ-Fe2O3-HBF4 were investigated using SEM and TEM. As shown in Fig. 6, the low magnification SEM images show small nano-sized grains having spherical and quasi spherical morphologies with a narrow size distribution, which indicates the nano-crystalline nature of the γ-Fe2O3 nanoparticles. The presence of some larger particles is attributed to aggregation or overlapping of smaller particles. The sizes of γ-Fe2O3-HBF4 were further analyzed using TEM and the results (Fig. 7) showed that the nanoparticles have nano-dimensions ranging from 10 to 20 nm. In the TEM images, the shapes are relatively rather rectangular, which is attributed to the presence of HBF4 groups physisorbed onto the γ-Fe2O3 surfaces.
 |
| | Fig. 6 (a) SEM of γ-Fe2O3. (b) SEM of γ-Fe2O3-HBF4. | |
 |
| | Fig. 7 (a) TEM of γ-Fe2O3. (b) TEM of γ-Fe2O3-HBF4. | |
Superparamagnetic particles are beneficial for magnetic separation, so the magnetic properties of γ-Fe2O3 and γ-Fe2O3-HBF4 were characterized using a VSM. The room temperature magnetization curves of γ-Fe2O3 and γ-Fe2O3-HBF4 are shown in Fig. 8. As expected, the bare γ-Fe2O3 showed the higher magnetic value (saturation magnetization, Ms) of 61.4 emu g−1; the Ms value of γ-Fe2O3-HBF4 is decreased due to the increasing amount of nonmagnetic material on the particle surface that makes a larger percentage of the particle mass nonmagnetic. However, this value is sufficiently high for magnetic separation. The strong magnetization of the nanoparticle was also revealed by simple attraction with an external magnet.
 |
| | Fig. 8 Magnetization curves of γ-Fe2O3 and γ-Fe2O3-HBF4. | |
The N2 adsorption–desorption isotherm provided a valuable tool for studying the textural and structure properties. The specific surface area of the powders was determined by use of the Brunauer–Emmett–Teller (BET) method. The BET results showed that the average surface area of γ-Fe2O3 was 73.79 m2 g−1 and that of γ-Fe2O3-HBF4 was 68.84 m2 g−1 (Fig. 9). It was noted that γ-Fe2O3 had a much higher surface area than γ-Fe2O3-HBF4. This seems logical as the successful anchoring of HBF4 on the surface of the MNPs decreases the surface area.
 |
| | Fig. 9 (a) N2 adsorption–desorption isotherm of γ-Fe2O3. (b) N2 adsorption–desorption isotherm of γ-Fe2O3-HBF4. | |
The one-pot synthesis of 12-substituted-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-diones 4 was achieved through the three-component condensation of 3,4-methylenedioxyaniline, aldehydes and 2-hydroxy-1,4-naphthoquinone in the presence of γ-Fe2O3-HBF4 as a heterogeneous catalyst (Scheme 1). To find the optimum conditions, the synthesis of 12-substituted-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4a) was used as a model reaction under a variety of different conditions. The effects of solvents and catalysts were evaluated for this reaction, and the results are summarized in Table 1. It was found that when the reaction was carried out in EtOH without any catalyst the yield of product was low (Table 1, entry 1). DMF as a solvent provided higher yields than other organic solvents (CH3CN, CHCl3, and toluene) (Table 1, entry 5 vs. entries 2–4). To improve the yield, we examined the reaction when using different catalysts. Some proton acids can catalyze this reaction with moderate yields. The best result was obtained when γ-Fe2O3-HBF4 was used, according to the yield and the reaction time. Therefore, γ-Fe2O3-HBF4 was chosen as the catalyst for this reaction. We also evaluated the amount of γ-Fe2O3-HBF4 required for this reaction. The results from Table 1 (entries 11–14) show that 10 mol% γ-Fe2O3-HBF4 at 130 °C in DMF is optimal for the reaction.
Table 1 Catalyst optimization for the synthesis 12-substituted-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-diones
| Entry |
Catalyst/mol% |
Solvent |
T/°C |
Time/h |
Yieldb/% |
| 100 mg mmol−1. Isolated yield. |
| 1 |
— |
EtOH |
Reflux |
10 |
5 |
| 2 |
— |
CH3CN |
Reflux |
5 |
13 |
| 3 |
— |
CHCl3 |
Reflux |
5 |
6 |
| 4 |
— |
Toluene |
Reflux |
3 |
32 |
| 5 |
— |
DMF |
130 |
3 |
54 |
| 6 |
Nano γ-Fe2O3 (100)a |
DMF |
130 |
3 |
58 |
| 7 |
p-TsOH (10) |
DMF |
130 |
1 |
76 |
| 8 |
SiO2-HBF4 (10) |
DMF |
130 |
1 |
81 |
| 9 |
FeCl3 (10) |
DMF |
130 |
2 |
59 |
| 10 |
Et3N (10) |
DMF |
130 |
3 |
67 |
| 11 |
γ-Fe2O3-HBF4 (10) |
DMF |
130 |
1 |
91 |
| 12 |
γ-Fe2O3-HBF4 (5) |
DMF |
130 |
1.5 |
80 |
| 13 |
γ-Fe2O3-HBF4 (15) |
DMF |
130 |
1 |
90 |
| 14 |
γ-Fe2O3-HBF4 (20) |
DMF |
130 |
1 |
91 |
In order to extend the above reaction (Scheme 1) to a library system, various kinds of arylaldehydes 2 (Table 2) were subjected to the reaction with 2-hydroxy-1,4-naphthoquinone 3 and 3,4-methylenedioxyaniline 1 to give the corresponding 12-substituted-benzo[h][1,3]dioxolo[4,5-b]-acridine-10,11-diones, and representative examples are shown in Table 2. All of the arylaldehydes 2 gave the expected products in high yields, whether bearing electron-withdrawing groups (such as halide and nitro) or electron-donating groups (such as an alkyl group), under the same reaction conditions. Therefore, we conclude that the electronic nature of the substitutents of the aromatic aldehydes had no significant effect on the reaction. Even heterocyclic aldehydes could be used in this reaction (Table 2, entries 16 and 17). In addition, an aliphatic aldehyde (Table 2, entry 18) also showed high reactivity under the standard conditions, providing the corresponding 12-methyl-5,10-dihydro-benzo[i][1,3]dioxolo[4,5-b]acridine-6,11-dione in a good yield of 72%. The structures of the ortho-quinones 4 are in full agreement with IR, 1H NMR, 13C NMR, and elemental analysis, as illustrated below for a representative example (compound 4a). Compound 4a exhibited characteristic IR stretching frequencies in the 1678 cm−1 region for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. In its 1H NMR spectrum, H-6 and H-9 occur as doublets, at 8.82 (J = 8.0 Hz) and 8.00 (J = 7.6 Hz) ppm respectively. H-6 occurs at about at 9 ppm, more downfield than expected of aromatic protons. This is explicable by the close proximity of these protons to the lone pairs of the neighbouring nitrogens and the consequent anisotropic and van de Waals deshielding. 13C NMR gave two very close carbonyl resonances at δ 180.1 and 179.9 ppm, and the very close chemical shifts for the carbonyl 13C NMR signals suggested an ortho-quinone structure. The molecular formulae of all the synthesized compounds were confirmed by elemental analysis. The purities of the compounds was ascertained by HPLC and were found to be >97% pure.
O. In its 1H NMR spectrum, H-6 and H-9 occur as doublets, at 8.82 (J = 8.0 Hz) and 8.00 (J = 7.6 Hz) ppm respectively. H-6 occurs at about at 9 ppm, more downfield than expected of aromatic protons. This is explicable by the close proximity of these protons to the lone pairs of the neighbouring nitrogens and the consequent anisotropic and van de Waals deshielding. 13C NMR gave two very close carbonyl resonances at δ 180.1 and 179.9 ppm, and the very close chemical shifts for the carbonyl 13C NMR signals suggested an ortho-quinone structure. The molecular formulae of all the synthesized compounds were confirmed by elemental analysis. The purities of the compounds was ascertained by HPLC and were found to be >97% pure.
Table 2 Preparation of 12-substituted-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-diones
| Entry |
R |
Time/h |
Product |
Yielda/% |
| Isolated yield. |
| 1 |
4-Cl-C6H4 |
1 |
4a |
91 |
| 2 |
2-Cl-C6H4 |
1.5 |
4b |
84 |
| 3 |
4-F-C6H4 |
1 |
4c |
89 |
| 4 |
2-F-C6H4 |
1.5 |
4d |
82 |
| 5 |
4-NO2-C6H4 |
1 |
4e |
92 |
| 6 |
4-MeO-C6H4 |
1 |
4f |
90 |
| 7 |
2-MeO-C6H4 |
1.5 |
4g |
85 |
| 8 |
3-MeO-C6H4 |
1 |
4h |
88 |
| 9 |
3-NO2-C6H4 |
1 |
4i |
84 |
| 10 |
3,4-(Cl)2-C6H4 |
1 |
4j |
80 |
| 11 |
2,4-(Cl)2-C6H4 |
1 |
4k |
82 |
| 12 |
2,5-(MeO)2-C6H3 |
1.5 |
4l |
79 |
| 13 |
3-Br-4-MeO-C6H3 |
1 |
4m |
89 |
| 14 |
3,5-(MeO)2-C6H3 |
1.5 |
4n |
80 |
| 15 |
3-F-4-MeO-C6H3 |
1 |
4o |
82 |
| 16 |
2-Furanyl |
1.5 |
4p |
86 |
| 17 |
2-Thiophenyl |
1.5 |
4q |
90 |
| 18 |
Me |
1 |
4r |
72 |
The formation of isomeric systems (ortho- and para-quinone units) is possible in the reaction. Therefore, we considered it desirable to obtain independent chemical evidence for the presence of ortho- or para-quinone units in 4. To this end, we reacted 4e with o-phenylenediamine for 30 min under solvent-free conditions, affording compound 5 in 99% yield, confirming the ortho-quinone structure (Scheme 3). The structure of 5 was fully characterized using spectroscopic data and elemental analysis. H-1 and H-4 occur as a multiplet at 9.25–9.41 ppm, more downfield than expected of aromatic protons. This is explicable by the close proximity of these protons to the lone pairs of the neighbouring nitrogens and the consequent anisotropic and van de Waals deshielding. The lack of any carbonyl signal in the 13C NMR spectrum of 5, and the fact that 5 is formed by the reaction of one molecule of 4e with one molecule of o-phenylenediamine, clearly support the structure of 5, which, in turn, further corroborates the structure of 4 and the regiochemistry of its formation.
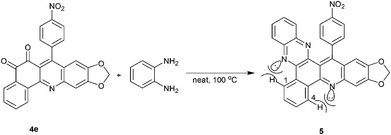 |
| | Scheme 3 Proof of the ortho-quinone structure of 4e, based on its reaction with o-phenylenediamine. | |
A plausible mechanism for the formation of the ortho-naphthoquinone is proposed in Scheme 4. We believe that the described transformations proceed via the initial formation of respective α,β-unsaturated carbonyl compounds, which undergo nucleophilic attack by the amine. This step is then followed by cyclization and oxidation to yield to product 4.
 |
| | Scheme 4 A plausible mechanistic pathway to explain the γ-Fe2O3-HBF4-catalyzed formation of 4. | |
The feasibility of the repeated use of γ-Fe2O3-HBF4 was also investigated for the reaction of 3,4-methylenedioxyaniline with 4-chloroaldehyde and 2-hydroxy-1,4-naphthoquinone. We found that the catalyst demonstrated excellent recyclability. The catalyst can be efficiently recovered easily and rapidly from the product by exposure to an external magnet (Fig. 10). To remove the residual product, the remaining magnetic nanoparticles were further washed with the EtOH, air-dried and used directly for the next round of reaction without further purification. The recycled catalyst was used for up to 6 runs with little loss of activity (Table 3).
 |
| | Fig. 10 Reaction mixture containing γ-Fe2O3-HBF4 (left), and γ-Fe2O3-HBF4 collected using an external magnet after the reaction (right). | |
Table 3 Recycling experiment for the synthesis of 4a
| Reaction cycles |
Elemental analysis of catalyst/% |
The recovery rate of catalyst/% |
Loading of HBF4a/mmol g−1 |
Yield/% |
| The amount of HBF4 loaded on the surface of nano γ-Fe2O3 was determined by ion-exchange pH analysis. |
| 1 |
B 0.35, F 2.43, H 0.065 |
96 |
0.32 |
91 |
| 2 |
B 0.32, F 2.24, H 0.057 |
92 |
0.29 |
89 |
| 3 |
B 0.31, F 2.14, H 0.056 |
90 |
0.28 |
87 |
| 4 |
B 0.30, F 2.07, H 0.056 |
90 |
0.27 |
84 |
| 5 |
B 0.29, F 1.99, H 0.055 |
86 |
0.26 |
82 |
| 6 |
B 0.27, F 1.90, H 0.051 |
85 |
0.25 |
79 |
The biological activities of this series of 12-substituted-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-diones were evaluated using a cytotoxicity assay, which was carried out with a panel of two human tumor cell lines (liver and ovarian) by using the MTT method.19 The results are summarized in Table 4, and are compared to doxorubicin. It is observed that most of the compounds are significantly cytotoxic. It appears from the data that 3-substitutions with an electron-withdrawing group in the 12-phenyl ring enhance the cytotoxicity for the two cancer cell lines, as seen in compounds 4o, 4i and 4m. A methyl substituent in the 12-phenyl ring also enhances the cytotoxicity (4r). Among the 19 new compounds screened, 4d has pronounced activity.
Table 4 Antitumor activities of compounds 4
| Compound |
IC50 (μM)a |
| HepG2 |
HeLa |
| The means of triplicates ± SD. |
| 4a |
32.83 ± 5.80 |
25.05 ± 5.51 |
| 4b |
>200 |
>200 |
| 4c |
>200 |
>200 |
| 4d |
4.81 ± 0.22 |
6.15 ± 0.77 |
| 4e |
90.89 ± 9.30 |
119.23 ± 8.87 |
| 4f |
>200 |
>200 |
| 4g |
29.48 ± 0.81 |
24.74 ± 4.84 |
| 4h |
>200 |
>200 |
| 4i |
13.08 ± 0.50 |
15.90 ± 4.52 |
| 4j |
>200 |
>200 |
| 4k |
>200 |
>200 |
| 4l |
82.31 ± 15.84 |
69.73 ± 7.04 |
| 4m |
27.63 ± 1.61 |
33.80 ± 2.04 |
| 4n |
>200 |
>200 |
| 4o |
5.91 ± 1.10 |
7.38 ± 3.08 |
| 4p |
>200 |
>200 |
| 4q |
>200 |
>200 |
| 4r |
8.59 ± 0.56 |
6.93 ± 0.47 |
| Doxorubicin |
2.25 ± 0.44 |
1.98 ± 0.33 |
Conclusion
In summary, we have synthesized the first γ-Fe2O3-HBF4 for use as a magnetic heterogeneous nanocatalyst. The catalyst is easily synthesized and can catalyze the synthesis of 12-substituted-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-diones with good to high yields. The characteristic aspects of this catalyst are rapid, simple and efficient separation by using an appropriate external magnet, which minimizes the loss of catalyst during separation, and the ability to reuse it several times with little loss of activity. In addition, this method is simple and convenient for the preparation of a wide range of ortho-quinone derivatives in a single-step operation, which are found to possess interesting antitumor properties.
Experimental section
General
IR spectra were obtained using a FTS-40 infrared spectrometer. NMR spectra were obtained using a Bruker AV-400 spectrometer at room temperature with TMS as the internal standard. Chemical shifts (δ) are given in ppm and coupling constants (J) in Hz. Elemental analysis was performed using a Vario-III elemental analyzer. Melting points were determined on a XT-4 binocular microscope and were uncorrected. Commercially available reagents were used throughout without further purification unless otherwise stated.
Large-scale preparation of magnetic γ-Fe2O3 nanoparticles
FeCl2·4H2O (9.25 mmol) and FeCl3·6H2O (15.8 mmol) were dissolved in deionized water (150 mL) under Ar atmosphere at room temperature. A NH4OH solution (25%, 50 mL) was then added dropwise to the stirring mixture at room temperature to change the reaction pH to 11. The resulting black dispersion was continuously stirred for 1 h at room temperature and then heated to reflux for 1 h to yield a brown dispersion. The magnetic nanoparticles were then purified using a repeated centrifugation, decantation, and redispersion cycle 3 times. The as-synthesized sample was heated at 2 °C min−1 up to 200 °C and then kept in the furnace for 3 h to give a reddish-brown powder.
Synthesis of γ-Fe2O3-HBF4
To a suspension of γ-Fe2O3 (16 g) in Et2O (500 mL), 40% aq. HBF4 (1.1 g, 5 mmol) was added. The mixture was agitated in an ultrasonic bath for 180 min at room temperature. The mixture was concentrated and the residue dried under vacuum at 100 °C for 72 h to afford γ-Fe2O3-HBF4 (0.32 mmol g−1).
Ion-exchange pH analysis
To an aqueous solution of NaCl (1 M, 25 mL) with a primary pH of 5.93, the catalyst (500 mg) was added and the resulting mixture was stirred for 2 h, after which the pH of the solution decreased to 2.19. This is equal to a loading of 0.32 mmol HBF4 g−1.
General procedure for the synthesis of 4
To a mixture of 3,4-methylenedioxyaniline (1 mmol), aldehyde (1 mmol), 2-hydroxy-1,4-naphthoquinone (1 mmol) and DMF (10 mL), γ-Fe2O3-HBF4 (0.1 mmol) was added. The mixture was stirred at 130 °C for an appropriate time (Table 2). After completion of the reaction, the catalyst was separated with the aid of an external magnet. Then the reaction mixture was cooled to room temperature. The precipitate was collected by filtration to afford the pure product 4.
12-(4-Chlorophenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4a). Orange powder, m.p. > 300 °C; IR (KBr): ν 3060, 2899, 1678, 1537, 1460, 1435, 1261, 1212, 1169, 1161, 1028, 944, 855, 772, 556 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.82 (d, 1H, J = 8.0 Hz, ArH), 8.00 (d, 1H, J = 7.6 Hz, ArH), 7.88 (t, 1H, J = 7.6 Hz, ArH), 7.64 (t, 1H, J = 7.6 Hz, ArH), 7.57 (d, 2H, J = 7.6 Hz, ArH), 7.51 (s, 1H, ArH), 7.26 (d, 2H, J = 7.6 Hz, ArH), 6.52 (s, 1H, ArH), 6.23 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 180.1, 179.9, 153.9, 150.1, 149.7, 149.6, 148.6, 137.7, 136.5, 135.9, 133.1, 132.3, 131.2, 130.5, 128.9, 128.5, 126.6, 124.8, 122.0, 106.1, 103.5, 102.1; anal. calc. for C24H12ClNO4: C 69.66, H 2.92, N 3.38; found: C 69.72., H 2.82, N 3.29.
12-(2-Chlorophenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4b). Orange powder, m.p. > 300 °C; IR (KBr): ν 3062, 2914, 1679, 1536, 1459, 1434, 1260, 1210, 1167, 1028, 944, 854, 771, 551 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.83 (d, 1H, J = 7.6 Hz, ArH), 8.01 (d, 1H, J = 8.0 Hz, ArH), 7.93–7.88 (m, 1H, ArH), 7.67–7.47 (m, 6H, ArH), 7.20 (dd, 1H, J = 1.6, 7.6 Hz, ArH), 6.36 (s, 1H, ArH), 6.27 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 179.4, 179.3, 154.1, 150.1, 150.0, 148.7, 147.8, 137.5, 136.6, 135.9, 132.3, 131.9, 131.3, 130.2, 130.0, 129.7, 128.7, 128.0, 126.5, 124.4, 121.8, 106.2, 103.7, 101.3; anal. calc. for C24H12ClNO4: C 69.66, H 2.92, N 3.38; found: C 69.60, H 3.02, N 3.33.
12-(4-Fluorophenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4c). Orange powder, m.p. > 300 °C; IR (KBr): ν 3067, 2902, 1681, 1608, 1538, 1462, 1434, 1260, 1225, 1207, 1170, 1031, 940, 861, 846, 557 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.82 (d, 1H, J = 7.6 Hz, ArH), 8.01 (d, 1H, J = 7.6 Hz, ArH), 7.89 (t, 1H, J = 7.2 Hz, ArH), 7.55 (s, 1H, ArH), 7.38–7.26 (m, 4H, ArH), 6.53 (s, 1H, ArH), 6.25 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 179.9, 179.8, 163.5, 153.8, 150.1, 149.9, 149.6, 148.4, 137.6, 135.8, 133.8, 132.3, 131.2, 130.7, 130.6, 128.5, 126.5, 125.0, 122.2, 115.9, 115.7, 106.1, 103.6, 102.1; anal. calc. for C24H12FNO4: C 72.54, H 3.04, N 3.52; found: C 72.58, H 3.00, N 3.61.
12-(2-Fluorophenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4d). Orange powder, m.p. 280–281 °C; IR (KBr): ν 3069, 2921, 1682, 1541, 1462, 1435, 1259, 1207, 1170, 1033, 942, 861, 769 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.83 (d, 1H, J = 7.6 Hz, ArH), 8.02 (d, 1H, J = 7.6 Hz, ArH), 7.89 (t, 1H, J = 7.6 Hz, ArH), 7.65 (t, 1H, J = 7.6 Hz, ArH), 7.59–7.55 (m, 2H, ArH), 7.39–7.35 (m, 2H, ArH), 7.24 (t, 1H, J = 7.2 Hz, ArH), 6.50 (s, 1H, ArH), 6.25 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 179.9, 179.6, 169.1, 154.1, 150.2, 150.0, 148.7, 144.7, 137.6, 135.9, 132.3, 131.2, 130.9, 130.8, 130.6, 130.5, 128.6, 125.0, 124.9, 116.0, 115.8, 106.2, 103.6, 101.5; anal. calc. for C24H12FNO4: C 72.54, H 3.04, N 3.52; found: C 72.58, H 3.00, N 3.61.
12-(4-Nitrophenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4e). Yellow powder, m.p. 299–300 °C; IR (KBr): ν 3057, 2914, 1682, 1593, 1540, 1520, 1462, 1435, 1352, 1258, 1166, 1036, 974, 859, 770, 555 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.83 (d, 1H, J = 7.6 Hz, ArH), 8.38 (d, 2H, J = 8.0 Hz, ArH), 8.02 (d, 1H, J = 7.6 Hz, ArH), 7.90 (t, 1H, J = 7.6 Hz, ArH), 7.65 (t, 1H, J = 7.6 Hz, ArH), 7.55–7.53 (m, 3H, ArH), 6.50 (s, 1H, ArH), 6.25 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 179.8, 179.6, 154.0, 150.1, 149.9, 148.7, 148.6, 147.2, 145.2, 137.6, 135.9, 132.4, 131.3, 130.1, 128.6, 126.6, 124.3, 124.0, 121.7, 106.2, 103.6, 101.9; MS (ESI): m/z 425 [M + H]+; anal. calc. for C24H12N2O6: C 67.93, H 2.85, N 6.60; found: C 68.01, H 2.81, N 6.57.
12-(4-Methoxyphenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4f). Orange red powder, m.p. > 300 °C; IR (KBr): ν 3066, 2957, 2838, 1679, 1611, 1536, 1460, 1434, 1260, 1250, 1167, 1028, 943, 852, 771, 559 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.81 (d, 1H, J = 8.0 Hz, ArH), 7.99 (d, 1H, J = 7.6 Hz, ArH), 7.88 (t, 1H, J = 7.6 Hz, ArH), 7.51 (s, 1H, ArH), 7.18–7.07 (m, 4H, ArH), 6.59 (s, 1H, ArH), 6.24 (s, 2H, OCH2O), 3.86 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 180.2, 180.0, 159.3, 153.6, 151.1, 150.1, 149.4, 148.4, 137.7, 135.8, 132.2, 131.1, 130.0, 129.5, 128.5, 126.6, 125.3, 122.3, 114.3, 106.0, 103.5, 102.3, 55.6; MS (ESI): m/z 410 [M + H]+; anal. calc. for C25H15NO5: C 73.35, H 3.69, N 3.42; found: C 73.42, H 3.62, N 3.32.
12-(2-Methoxyphenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4g). Orange red powder, m.p. > 300 °C; IR (KBr): ν 3061, 2953, 2841, 1681, 1536, 1463, 1434, 1257, 1237, 1166, 1035, 945, 756 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.81 (d, 1H, J = 8.0 Hz, ArH), 7.99 (d, 1H, J = 7.6 Hz, ArH), 7.89 (t, 1H, J = 7.6 Hz, ArH), 7.64 (t, 1H, J = 7.6 Hz, ArH), 7.52 (s, 1H, ArH), 7.50–7.47 (m, 1H, ArH), 7.19 (d, 1H, J = 8.4 Hz, ArH), 7.10–7.00 (m, 2H, ArH), 6.46 (s, 1H, ArH), 6.24 (s, 2H, OCH2O), 3.81 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 179.9, 179.8, 156.6, 153.9, 132.2, 131.1, 130.0, 129.3, 128.6, 126.5, 126.3, 125.1, 122.3, 121.1, 111.8, 106.0, 103.5, 102.0, 55.9; anal. calc. for C25H15NO5: C 73.35, H 3.69, N 3.42; found: C 73.32, H 3.75, N 3.53.
12-(3-Methoxyphenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4h). Orange powder, m.p. > 300 °C; IR (KBr): ν 3059, 2921, 1682, 1538, 1462, 1434, 1259, 1231, 1165, 1033, 947, 846, 768 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.82 (d, 1H, J = 8.0 Hz, ArH), 8.00 (d, 1H, J = 7.6 Hz, ArH), 7.88 (t, 1H, J = 7.6 Hz, ArH), 7.64 (t, 1H, J = 7.6 Hz, ArH), 7.50 (s, 1H, ArH), 7.44 (t, 1H, J = 7.6 Hz, ArH), 7.06 (d, 1H, J = 8.0 Hz, ArH), 6.80 (s, 2H, ArH), 6.55 (s, 1H, ArH), 6.23 (s, 2H, OCH2O), 3.84 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 180.1, 180.0, 159.9, 153.8, 150.9, 150.1, 149.5, 148.5, 139.0, 137.8, 135.9, 132.3, 131.1, 130.0, 128.5, 126.6, 125.0, 121.9, 120.8, 114.4, 113.7, 106.0, 103.5, 102.3, 55.7; anal. calc. for C25H15NO5: C 73.35, H 3.69, N 3.42; found: C 73.40, H 3.59, N 3.47.
12-(3-Nitrophenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4i). Yellow powder, m.p. > 300 °C; IR (KBr): ν 3069, 2917, 1685, 1534, 1462, 1434, 1351, 1258, 1208, 1168, 1031, 945, 862, 741 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.84 (d, 1H, J = 8.0 Hz, ArH), 8.36 (d, 1H, J = 8.0 Hz, ArH), 8.12 (s, 1H, ArH), 8.02 (d, 1H, J = 7.6 Hz, ArH), 7.91–7.82 (m, 2H, ArH), 7.72 (d, 1H, J = 7.6 Hz, ArH), 7.65 (t, 1H, J = 7.2 Hz, ArH), 7.55 (s, 1H, ArH), 6.55 (s, 1H, ArH), 6.245 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 179.6, 179.5, 153.9, 150.0, 149.9, 148.5, 148.0, 139.5, 137.5, 135.9, 135.4, 132.3, 131.3, 130.5, 128.6, 126.5, 124.6, 123.5, 132.2, 122.1, 117.7, 106.1, 103.6, 102.0; anal. calc. for C24H12N2O6: C 67.93, H 2.85, N 6.60; found: C 67.88, H 2.88, N 6.52.
12-(3,4-Dichlorophenyl)-benzo[h][1,3]dioxolo[4,5-b]-acridine-10,11-dione (4j). Yellow powder, m.p. > 300 °C; IR (KBr): ν 3065, 2922, 1683, 1612, 1537, 1463, 1435, 1260, 1209, 1168, 1028, 943, 853, 768 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.80 (d, 1H, J = 8.0 Hz, ArH), 8.01 (d, 1H, J = 8.0 Hz, ArH), 7.88 (t, 1H, J = 7.6 Hz, ArH), 7.79 (d, 1H, J = 8.0 Hz, ArH), 7.64 (t, 1H, J = 7.6 Hz, ArH), 7.54–7.52 (m, 2H, ArH), 7.25 (dd, 1H, J = 2.0 and 8.0 Hz, ArH), 6.60 (s, 1H, ArH), 6.26 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 176.6, 176.5, 153.9, 149.9, 149.8, 148.5, 147.8, 138.5, 137.5, 135.9, 132.2, 131.7, 131.2, 131.0, 130.5, 129.0, 128.6, 126.5, 124.5, 122.0, 106.1, 103.6, 102.0; anal. calc. for C24H11Cl2NO4: C 64.31, H 2.47, N 3.12; found: C 64.24, H 2.53, N 3.19.
12-(2,4-Dichlorophenyl)-benzo[h][1,3]dioxolo[4,5-b] acridine-10,11-dione (4k). Yellow powder, m.p. > 300 °C; IR (KBr): ν 3060, 2918, 1680, 1538, 1464, 1436, 1260, 1209, 1171, 1033, 943, 855, 794 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.82 (d, 1H, J = 8.0 Hz, ArH), 8.02 (d, 1H, J = 7.6 Hz, ArH), 7.90 (t, 1H, J = 7.6 Hz, ArH), 7.79 (s, 1H, ArH), 7.65 (t, 1H, J = 7.6 Hz, ArH), 7.58–7.54 (m, 2H, ArH), 7.26 (d, 1H, J = 8.4 Hz, ArH), 6.470 (s, 1H, ArH), 6.27 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 179.6, 179.5, 154.3, 150.2, 150.1, 148.9, 146.7, 137.5, 136.0, 135.7, 134.0, 133.1, 132.3, 131.4, 131.3, 129.4, 128.7, 128.2, 126.5, 124.3, 121.8, 106.3, 103.7, 101.4; anal. calc. for C24H11Cl2NO4: C 64.31, H 2.47, N 3.12; found: C 64.35, H 2.45, N 3.13.
12-(2,5-Dimethoxyphenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4l). Orange powder, m.p. > 300 °C; IR (KBr): ν 3073, 3002, 2961, 2837, 1681, 1670, 1540, 1492, 1463, 1436, 1261, 1228, 1217, 1166, 1039, 1017, 948, 864, 729 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.83 (d, 1H, J = 8.4 Hz, ArH), 8.01 (d, 1H, J = 7.2 Hz, ArH), 7.90 (t, 1H, J = 7.6 Hz, ArH), 7.64 (t, 1H, J = 7.6 Hz, ArH), 7.51 (s, 1H, ArH), 7.13–7.03 (m, 2H, ArH), 6.64 (s, 1H, ArH), 6.54 (s, 1H, ArH), 6.24 (s, 2H, OCH2O), 3.72 (s, 3H, OCH3), 3.55 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 180.2, 180.0, 154.0, 153.9, 150.7, 150.1, 149.6, 148.6, 137.8, 136.0, 132.2, 131.2, 128.6, 127.3, 126.6, 126.0, 125.0, 122.3, 115.5, 114.6, 113.3, 106.0, 103.5, 102.1, 56.6, 56.1; anal. calc. for C26H17NO6: C 71.07, H 3.90, N 3.19; found: C 71.23, H 3.87, N 3.21.
12-(3,5-Dimethoxyphenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4m). Orange red powder, m.p. > 300 °C; IR (KBr): ν 3095, 2968, 2844, 1681, 1594, 1536, 1462, 1435, 1259, 1208, 1162, 1029, 935, 860, 768 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.82 (d, 1H, J = 7.2 Hz, ArH), 8.01 (d, 1H, J = 8.0 Hz, ArH), 7.91–7.87 (m, 1H, ArH), 7.66–7.63 (m, 1H, ArH), 7.53 (s, 1H, ArH), 6.62 (s, 1H, ArH), 6.37 (d, 1H, J = 2.4 Hz, ArH), 6.25 (s, 2H, OCH2O), 3.75 (s, 6H, 2OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 179.8, 179.6, 160.0, 153.8, 150.7, 150.0, 149.5, 148.3, 139.7, 137.7, 135.8, 132.3, 131.1, 128.5, 126.5, 124.8, 121.9, 106.6, 106.0, 103.5, 102.2, 99.8, 55.8; anal. calc. for C26H17NO6: C 71.07, H 3.90, N 3.19; found: C 71.15, H 3.96, N 3.16.
12-(3-Bromo-4-methoxyphenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4n). Orange powder, m.p. > 300 °C; IR (KBr): ν 3059, 2061, 1682, 1611, 1536, 1461, 1435, 1287, 1257, 1241, 1206, 1169, 1030, 941, 767 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.81 (d, 1H, J = 8.0 Hz, ArH), 8.00 (d, 1H, J = 8.0 Hz, ArH), 7.88 (t, 1H, J = 7.2 Hz, ArH), 7.64 (t, 1H, J = 7.2 Hz, ArH), 7.53–7.45 (m, 2H, ArH), 7.26–7.223 (m, 2H, ArH), 6.60 (s, 1H, ArH), 6.24 (s, 2H, OCH2O), 3.97 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 180.2, 180.1, 155.6, 153.8, 149.7, 149.3, 148.6, 137.7, 135.9, 132.9, 132.2, 131.1, 131.0, 129.4, 129.3, 126.6, 125.9, 125.2, 122.2, 113.2, 111.2, 106.1, 103.5, 102.2, 56.9; anal. calc. for C25H14BrNO5: C 61.49, H 2.89, N 2.87; found: C 61.55, H 3.00, N 2.81.
12-(3-Fluoro-4-methoxyphenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4o). Orange red powder, m.p. > 300 °C; IR (KBr): ν 3063, 2920, 2851, 1682, 1538, 1464, 1434, 1321, 1259, 1211, 1166, 1034, 940, 854, 760 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.80 (d, 1H, J = 8.0 Hz, ArH), 8.00 (d, 1H, J = 7.6 Hz, ArH), 7.88 (t, 1H, J = 8.0 Hz, ArH), 7.64 (t, 1H, J = 7.6 Hz, ArH), 7.52 (s, 1H, ArH), 7.31 (t, 1H, J = 8.0 Hz, ArH), 7.13 (d, 1H, J = 11.2 Hz, ArH), 7.00 (d, 1H, J = 8.0 Hz, ArH), 6.63 (s, 1H, ArH), 6.25 (s, 2H, OCH2O), 3.95 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6) δ: 179.9, 179.8, 167.4, 153.7, 150.7, 149.9, 149.6, 148.4, 147.2, 137.6, 135.8, 132.2, 131.2, 128.5, 126.5, 125.1, 125.0, 122.3, 116.7, 116.5, 114.3, 106.0, 103.6, 102.2, 56.5; anal. calc. for C25H14FNO5: C 70.26, H 3.30, N 3.28; found: C 70.33, H 3.25, N 3.24.
12-((Furan-2-yl))-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4p). Orange red powder, m.p. > 300 °C; IR (KBr): ν 3060, 2953, 1679, 1595, 1535, 1460, 1433, 1261, 1225, 1166, 1030, 945, 771, 592 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.76 (d, 1H, J = 7.6 Hz, ArH), 8.00 (d, 1H, J = 8.0 Hz, ArH), 7.91 (s, 1H, ArH), 7.87 (t, 1H, J = 7.6 Hz, ArH), 7.64 (t, 1H, J = 7.6 Hz, ArH), 7.52 (s, 1H, ArH), 7.02 (s, 1H, ArH), 6.75–6.69 (m, 2H, ArH), 6.29 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 179.7, 179.6, 154.1, 150.2, 148.8, 147.6, 144.7, 138.5, 137.4, 135.8, 132.3, 131.2, 128.5, 126.4, 124.5, 122.8, 111.9, 111.8, 106.2, 103.7, 101.5; anal. calc. for C22H11NO5: C 71.55, H 3.00, N 3.79; found: C 71.60, H 2.96, N 3.84.
12-(Thiophen-2-yl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4q). Orange red powder, m.p. > 300 °C; IR (KBr): ν 3015, 2924, 1679, 1542, 1460, 1434, 1260, 1205, 1160, 1034, 941, 859, 723 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.82 (d, 1H, J = 7.6 Hz, ArH), 8.01 (d, 1H, J = 7.6 Hz, ArH), 7.91–7.87 (m, 2H, ArH), 7.88 (t, 1H, J = 7.6 Hz, ArH), 7.66–7.62 (m, 1H, ArH), 7.53 (s, 1H, ArH), 7.24 (s, 1H, ArH), 7.03 (s, 1H, ArH), 6.75 (s, 1H, ArH), 6.26 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 179.9, 179.7, 154.0, 150.3, 148.5, 147.2, 144.4, 137.0, 135.9, 132.3, 131.2, 128.5, 127.9, 127.7, 126.6, 126.1, 123.2, 113.5, 113.0, 105.9, 103.6, 101.8; anal. calc. for C22H11NO4S: C 68.56, H 2.88, N 3.63; found: C 68.55, H 2.92, N 3.70.
12-Methyl-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (4r). Orange red powder, m.p. 263–265 °C; IR (KBr): ν 3066, 2922, 1679, 1601, 1571, 1501, 1469, 1437, 1253, 1243, 1037, 939, 774, 721 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 8.71 (d, 1H, J = 8.0 Hz, ArH), 7.92 (d, 1H, J = 6.8 Hz, ArH), 7.76 (t, 1H, J = 7.6 Hz, ArH), 7.63 (t, 1H, J = 7.6 Hz, ArH), 7.48 (s, 1H, ArH), 6.29 (s, 2H, OCH2O), 1.22 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ: 179.1, 178.9, 154.3, 149.2, 148.2, 147.3, 145.4, 137.1, 135.3, 132.4, 131.0, 128.9, 126.5, 125.7, 118.0, 108.9, 104.7, 102.0, 24.7; anal. calc. for C19H11NO4: C 71.92, H 3.49, N 4.41; found: C 72.02, H 3.44, N 4.37.
Typical procedure for the synthesis of compound 5
A mixture of 12-(4-nitrophenyl)-benzo[h][1,3]dioxolo[4,5-b]acridine-10,11-dione (1 mmol) and o-phenylenediamine (1.2 mmol) was heated at 100 °C for an appropriate time and monitored by TLC until conversion was complete. The reaction mixture was then cooled to room temperature and diluted with cold water (40 mL). The solid product was collected by filtration and was purified by recrystallization from 95% EtOH to afford the desired pure product 5 as a pale yellow solid.
12-(4-Nitrophenyl)-benzo[h][1,3]dioxolo[4,5-b]-quinoxalo-[2,3-j]acridine-10,11-dione (5). Yellow powder, m.p. > 300 °C; IR (KBr): ν 3057, 2910, 1512, 1497, 1461, 1439, 1345, 1270, 1213, 1183, 1041, 972, 856, 774, 552 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 9.41 (d, 1H, J = 7.6 Hz, ArH), 9.25 (d, 2H, J = 7.6 Hz, ArH), 8.50 (d, 2H, J = 8.4 Hz, ArH), 8.20 (d, 1H, J = 8.4 Hz, ArH), 7.88–7.74 (m, 3H, ArH), 7.65–7.52 (m, 4H, ArH), 7.09 (d, 1H, J = 8.4 Hz, ArH), 6.70 (s, 1H, ArH), 6.16 (s, 2H, OCH2O); 13C NMR (100 MHz, DMSO-d6) δ: 152.1, 149.1, 148.8, 147.5, 147.2, 147.0, 143.0, 142.3, 141.0, 140.1, 131.8, 130.7, 130.3, 130.0, 129.9, 129.8, 129.1, 128.8, 125.9, 125.4, 120.8, 119.1, 105.7, 102.2, 101.2; anal. calc. for C30H16N4O4: C 72.58, H 3.25, N 11.28; found: C 72.62, H 3.30, N 11.19.
Cytotoxicity assay
Cell viability for all cell lines was determined using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) colorimetric assay. Compounds 4a–4r, 5 were subjected to cytotoxic evaluation against HeLa and HepG2 cell lines employing the colorimetric method. Doxorubicin was used as the reference substance.
MTT was dissolved in saline to make a concentration of 5 mg μL−1 as a stock solution. Cancer cells (3 × 103 cells) suspended in 100 mg per well of MEM medium containing 10% fetal calf serum were seeded onto a 96-well culture plate. After 24 h pre-incubation at 37 °C in a humidified atmosphere of 5% CO2/95% air to allow cell attachment, various concentrations of test solution (10 μL per well) were added and then incubated for 48 h under the above conditions. At the end of the incubation, 10 μL of tetrazolium reagent was added into each well and then incubated at 37 °C for 4 h. The supernatant was decanted, and DMSO (100 μL per well) was added to allow formazan solubilization. The optical density (OD) of each well was detected using a microplate reader at 550 nm and for correction at 595 nm. Each determination represents the average means of six replicates. The 50% inhibition concentration (IC50) was determined by curve fitting.
Acknowledgements
We are pleased to acknowledge the financial support from Xinxiang University.
References
-
(a) K. Nepali, S. Sharma, M. Sharma, P. M. S. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422 CrossRef CAS PubMed;
(b) K. Nepali, S. Sharma, D. Kumar, A. Budhiraja and K. L. Dhar, Recent Pat. Anti-Cancer Drug Discovery, 2014, 9, 303 CAS.
- J. P. Kim, W. G. Kim, H. Koshino, J. Jung and I. Yoo, Phytochemistry, 1996, 43, 425 CrossRef CAS.
-
(a) G. Errante, G. L. Motta, C. Lagana, V. Wittebolle, M.-E. Sarciron and R. Barret, Eur. J. Med. Chem., 2006, 41, 773 CrossRef CAS PubMed;
(b) S. Gafner, J. L. Wolfender, M. Nianga, H. Stoeckli-Evans and K. Hostettmann, Phytochemistry, 1996, 42, 1315 CrossRef CAS.
- D. O. Moon, Y. H. Choi, N. D. Kim, Y. M. Park and G. Y. Kim, Int. Immunopharmacol., 2007, 7, 506 CrossRef CAS PubMed.
- V. K. Tando, R. V. Singhb and D. B. Yadava, Bioorg. Med. Chem. Lett., 2004, 14, 2901 CrossRef PubMed.
- M. Yamashita, M. Kaneko, A. Iida, H. Tokudab and K. Nishimura, Bioorg. Med. Chem. Lett., 2007, 17, 6417 CrossRef CAS PubMed.
- L. Zhou, W. K. Chan, N. Xu, K. Xiao, H. Luo, K. Q. Luo and D. C. Chang, Life Sci., 2008, 83, 394 CrossRef CAS PubMed.
- X. Chen, J. Ding, Y. M. Ye and J. S. Zhang, J. Nat. Prod., 2002, 65, 1016 CrossRef CAS PubMed.
- M. Dubin, S. H. Fernandez Villamil and A. O. Stoppani, Medicina, 2001, 61, 343 CAS.
- D. Wang, M. Y. Xia, Z. Cui, S. I. Tashiro, S. Onodera and T. Ikejima, Biol. Pharm. Bull., 2004, 27, 1025 CAS.
- J. S. Zhang, J. Ding, Q. M. Tang, M. Li, M. Zhao, L. J. Lu, L. J. Chen and S. T. Yuan, Bioorg. Med. Chem. Lett., 1999, 9, 2731 CrossRef CAS.
- C. Qing, C. Jiang, J. S. Zhang and J. Ding, Anticancer Drugs, 2001, 12, 51 CrossRef CAS PubMed.
-
(a) H. R. Lu, L. H. Meng, M. Huang, H. Zhu, Z. H. Miao and J. Ding, Cancer Chemother. Pharmacol., 2005, 55, 286 CrossRef CAS PubMed;
(b) Z. H. Miao, T. Tang, Y. X. Zhang, J. S. Zhang and J. Ding, Int. J. Cancer, 2003, 106, 108 CrossRef CAS PubMed.
- L. H. Meng, J. S. Zhang and J. Ding, Biochem. Pharmacol., 2001, 62, 733 CrossRef CAS.
-
(a) A. H. Lu, E. L. Salabas and F. Schueth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed;
(b) R. Fu, Xi. Jin, J. Liang, W. Zheng, J. Zhuang and W. Yang, J. Mater. Chem., 2011, 21, 15352 RSC;
(c) M. Niu, M. Du, Z. Gao, C. Yang, X. Lu, R. Qiao and M. Gao, Macromol. Rapid Commun., 2010, 31, 1805 CrossRef CAS PubMed;
(d) S. Sobhani, Z. P. Parizi and N. Razavi, Appl. Catal., A, 2011, 409–410, 162 CrossRef CAS PubMed.
-
(a) A. Megia-Fernandez, M. Ortega-Munoz, J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Adv. Synth. Catal., 2010, 352, 3306 CrossRef CAS;
(b) B. G. Wang, B. C. Ma, Q. O. Wang and W. Wang, Adv. Synth. Catal., 2010, 352, 2923 CrossRef CAS;
(c) H. Firouzabadi, N. Iran-poor, M. Gholinejad and J. Hoseini, Adv. Synth. Catal., 2011, 353, 125 CrossRef CAS;
(d) A. J. Amali and R. K. Rana, Green Chem., 2009, 11, 1781 RSC;
(e) R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2010, 12, 144 RSC.
-
(a) J.-P. Zhang, W. Fan, J. Ding, B. Jiang, S.-J. Tu and G. Li, Heterocycles, 2014, 88, 1065 CrossRef CAS PubMed;
(b) S. Mahajan, S. Khullar, S. K. Mandal and I. P. Singh, Chem. Commun., 2014, 50, 10078 RSC;
(c) A. Schatz, O. Reiser and W. J. Stark, Chem.–Eur. J., 2010, 16, 8950 CrossRef PubMed.
- L. Q. Wu and Z. K. Yin, Eur. J. Inorg. Chem., 2013, 6156 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00887e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 













