
Chemical synthesis of the tumor-associated globo H antigen†
Abstract
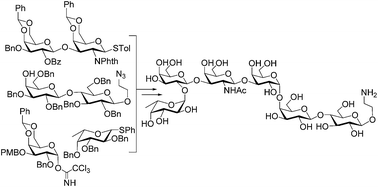
A derivative of the tumor-associated globo H antigen, a complex hexasaccharide, was synthesized by a convergent and efficient [3 + 2 + 1] strategy using various glycosylation methods. All glycosylation reactions afforded good to excellent yields and outstanding stereoselectivity, including the installation of cis α-linked D-galactose and L-fucose. The longest linear sequence for this synthesis was 11 steps from a galactose derivative 11 to give an overall yield of 2.6%. The synthetic target had a free and reactive amino group at the glycan reducing end, facilitating its conjugation with other molecules for various applications.
 Please wait while we load your content...
Please wait while we load your content...

