
Low-concentration methane combustion over a Cu/γ-Al2O3 catalyst: effects of water
Haojie Geng,
Zhongqing Yang*,
Jingyu Ran,
Li Zhang,
Yunfei Yan and
Mingnv Guo
Key Laboratory of Low-Grade Energy Utilization Technologies and Systems, Ministry of Education, College of Power Engineering, Chongqing University, Shapingba District, Chongqing 400030, People’s Republic of China. E-mail: zqyang@cqu.edu.cn; Fax: +86-23-65111832; Tel: +86-023-65103114
First published on 3rd February 2015
Abstract
The influence of water on low-concentration methane oxidation over a Cu/γ-Al2O3 catalyst was investigated in a fixed bed reactor. This paper studied the effect of water on the activity of methane combustion, using parameters such as water reversible adsorption, regeneration of the activity and surface characteristics of the catalyst. Apparent activation energy was found by experiments, and water surface coverage was calculated using the Langmuir equation. It was found that the activity of methane combustion over a Cu/γ-Al2O3 catalyst decreased with time due to water adsorption. The inhibitory effect generated by water weakened as the temperature rose above 550 °C. Reactivity could be refreshed if the catalyst particles were scavenged by N2. Kinetic experiments showed that, if water was added into the feed, apparent activation energy (Ea) increased noticeably (81.4 kJ mol−1 → 153.0 kJ mol−1) and the reaction order with respect to water was −0.6 to −1. Using the Langmuir equation, it could be concluded that the coverage of water adsorption on catalytic active sites increased noticeably as vapor was introduced into the feed. If the temperature increased, water coverage went down and tended towards 0% above 625 °C.
1. Introduction
There was a large amount of low-concentration methane in the coal bed, especially in China. It was hard to make use of the methane using conventional combustion technology and there was no choice but to emit the gas into the atmosphere. Catalytic combustion had been considered to be a comparatively ideal burning mode so far, which could achieve high-conversion-efficiency and low-pollution-emission with noble metal or transition metal catalysts.1–4 A large number of studies had been done on the catalytic combustion of methane with noble or transition metal catalysts, and combustion mechanism had been widely reported.5–7 However the feed always contained a large amount of steam which could affect the reactivity during the actual reaction. Therefore, the effect of water on methane combustion required further research.8–10Many catalysts had been investigated for their activity in the catalytic combustion of methane with water over noble metal catalysts (such as Pd, Pt) and many conclusions were drawn. Some scholars considered that H2O adsorbed on the active sites, which formed OH* groups, generated inhibitory effects on methane combustion.11–15 Among the combustion products, H2O had a more obvious influence than CO2 on methane combustion. In oxygen-rich conditions (O2/CH4 > 2), it was hard to progress the reforming reaction of methane with water. About the influence of water, the studies of Ciuparu et al.16,17 reported that, by means of in situ DR-FTIR investigation, several OH adsorption bands were observed on the catalyst surface and hydroxyls were found to be related to the PdO phase. OH groups had bridged and terminal bonds before recombination and desorption as water molecules, suggesting the dehydroxylation mechanism proceeds. Stasinska et al.18,19 reported that steam could promote the growth of surface grain, resulting in the decrease of the surface area of the catalyst. Meanwhile, Qiao et al.20,21 reported that water vapor could severely inhibit the catalytic performance of CeO2–CuO solid solutions for CH4 and CO conversion, while CeO2–NiO showed a good resistance to water. Using special treatments to ensure differential conditions, J. C van Giezen22 reported that apparent activation energy of methane (Ea) increased from 86 kJ mol−1 to 151 kJ mol−1 if water was introduced into the feed and the order with respect to water was found to be −0.76.
For methane combustion with water (O2/CH4 > 2), the inhibitory effects of water had been widely acknowledged, especially in Pd or Pt.23–26 However, there were few studies about transition metal catalysts, such as copper. The inhibitory effects of water at low temperature, adsorption characteristics and catalyst surface property were not yet clear for methane combustion. H2O surface coverage with differing temperature required further study. The aim of this work was to investigate the inhibitory effects of water on the copper based catalyst from the following aspects: water adsorption, regeneration of activity, kinetic parameters of methane and water surface coverage.
2. Experimental
2.1. Catalyst preparation and characterization
γ-Al2O3 particles (specific surface area, 181.4 m2 g−1; pore volume, 0.46 cm3 g−1; pore diameter, 190.6 Å) were used as supports of the Cu catalyst. The γ-Al2O3 particle diameter was lower than 0.1 mm, and each was calcined at 500 °C for 6 h before loading. Alumina powder was impregnated with the metal using the incipient wetness technique. 0.294 g Cu(NO3)2 powder was chosen to load 0.1 g Cu on the support (1 g). The mass of distilled water, the solvent for the Cu(NO3)2 powder, was determined by the support mass and pore volume. Support particles were impregnated with Cu(NO3)2 solution for 5 h; after impregnation, the catalyst was dried at 150 °C in a drying oven for 6 h in air and calcined for 6 h in a muffle furnace at 500 °C in N2. The self-made catalyst was cooled down to room temperature in a muffle furnace (under N2 atmosphere). During this process, the nitrate precursor and hydroxyl group were completely decomposed and desorbed fully from the catalyst. Table 1 and 2 show the content of each element (XRF) and specific surface area (BET) of the self-made catalyst.| Cu | Al | O | Si | K | Other | |
|---|---|---|---|---|---|---|
| Mass (wt/%) | 9.64% | 40.76% | 42.98% | 1.36% | 1.78% | 3.84% |
| Specific surface area | Pore volume | Pore diameter |
|---|---|---|
| 180.6 m2 g−1 | 0.4571 cm3 g−1 | 188.363 Å |
Fig. 1 shows the XRD spectra of the Cu/γ-Al2O3 catalyst: a, fresh catalyst; b, catalyst after reaction. From Fig. 1(a) and (b), the fresh catalyst consisted of CuO and Al2O3 between 30° and 70°. Other species (Cu1+, O1−, O2 ads) were not found in Fig. 1. This meant that CuO was the active site that provided O* (chemisorbed O) to methane. In the flow reactor, as O2 was enough for CH4 oxidation (O2/CH4 > 2), the Cu cluster, especially surface Cu, was able to maintain Cu2+ and provide O*. Fig. 1(b) showed that, during the reaction, the catalyst phase did not change much, and other peaks were not presented after methane oxidation.
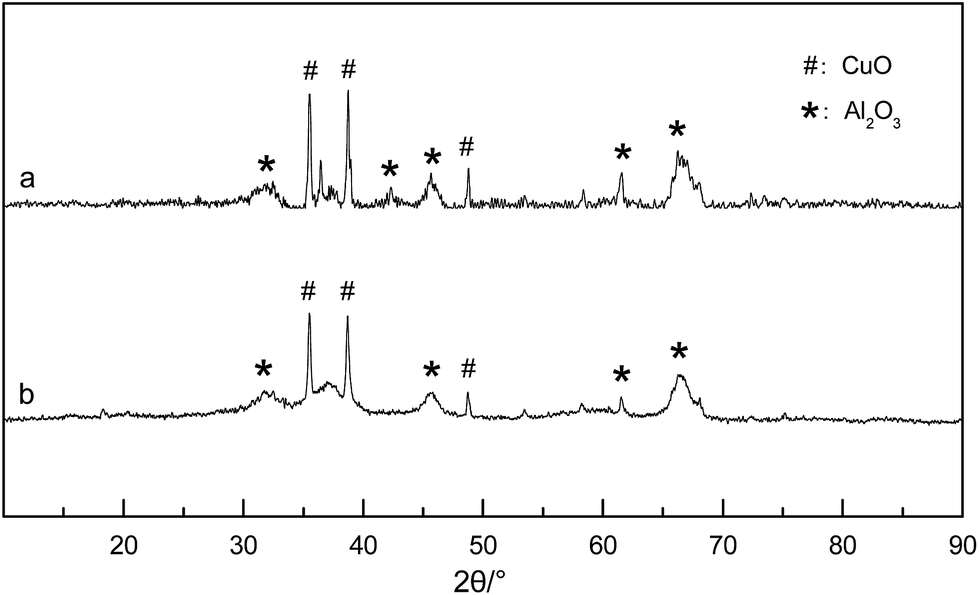 | ||
| Fig. 1 XRD spectra of the Cu/γ-Al2O3 catalyst. (a) Fresh catalyst; (b) catalyst after reaction (550 °C, 3 vol% CH4, 20 vol% O2, N2 balance). | ||
2.2. Experimental devices and system
Fig. 2 presents the experimental device and system. The experiment of methane combustion was conducted in the fixed-bed reactor which had a ceramic reaction tube with 10 mm inner diameter and 1200 mm length. The reaction mixture gas (3 vol% CH4, 20 vol% O2 and N2 balance) controlled by a mass flowmeter, was injected into the fixed-bed reactor at 200 ml min−1. Water vapor, generated by a vapor generator (W-202A-220-K) was carried by N2 at 150 °C and mixed with CH4 and O2 before entering into the reactor. The reactor was heated by a resistance-heated furnace that was connected to the data acquisition and control system which controlled the reaction temperature. The effluent gas was measured by gas chromatography to determine the concentrations of CH4 and CO2.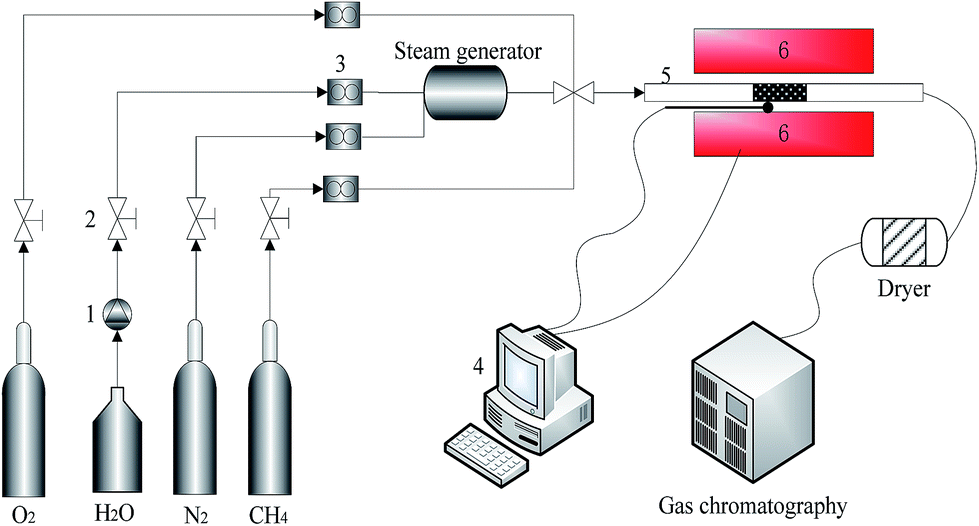 | ||
| Fig. 2 Flow chart of the experimental system; 1. Flow control pump; 2. pressure relief valve; 3. mass flowmeter; 4. data acquisition and control system; 5. fixed-bed reactor; 6. resistance stove. | ||
2.3. Catalytic activity test for methane oxidation
The reactivity of Cu/γ-Al2O3 was determined by methane conversion. According to the carbon balance, methane conversion x is described as:
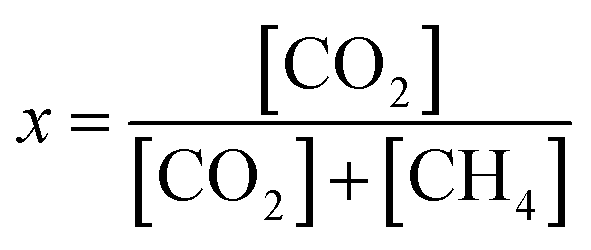 | (1) |
The microstructure and characterization of the catalyst surface were investigated by scanning electron microscopy (SEM), which magnified the catalyst surface by 1000 times. BET surface area, pore volume and pore diameter were measured by the BET method before or after the experiments.
3. Results and discussion
3.1. Effects of water on methane combustion
The effect of water on methane combustion over a Cu/γ-Al2O3 catalyst is shown in Fig. 3 and 4. Fig. 3 shows the change in methane conversion with temperature at different water content values (0 vol%, 2.5 vol%, 5 vol%, 10 vol%, 15 vol%, 20 vol%). Fig. 4 shows the inhibitory effect of water on methane combustion at 500 °C and 600 °C. From these figures, water significantly affected catalytic activity and CH4 conversion dropped if water was introduced into the feed. When the water vapor increased from 0 vol% to 10 vol%, the catalyst continued losing activity with time. From Fig. 3, with temperature rising from 475 °C to 600 °C, CH4 conversion increased quickly. The inhibition effect of H2O on methane combustion increased firstly and then decreased, and water inhibition among the curves reached its maximum at 525 °C. The process showed that water vapor had a significant inhibitory effect on methane combustion, while, with temperature increasing, the influence weakened gradually. A considerably higher temperature was most likely required for the catalytic reaction to proceed. | ||
| Fig. 3 Methane oxidation with water over a Cu/γ-Al2O3 catalyst (CH4, 3 vol%; O2, 20 vol%; H2O, 0–20 vol%; N2, balance). | ||
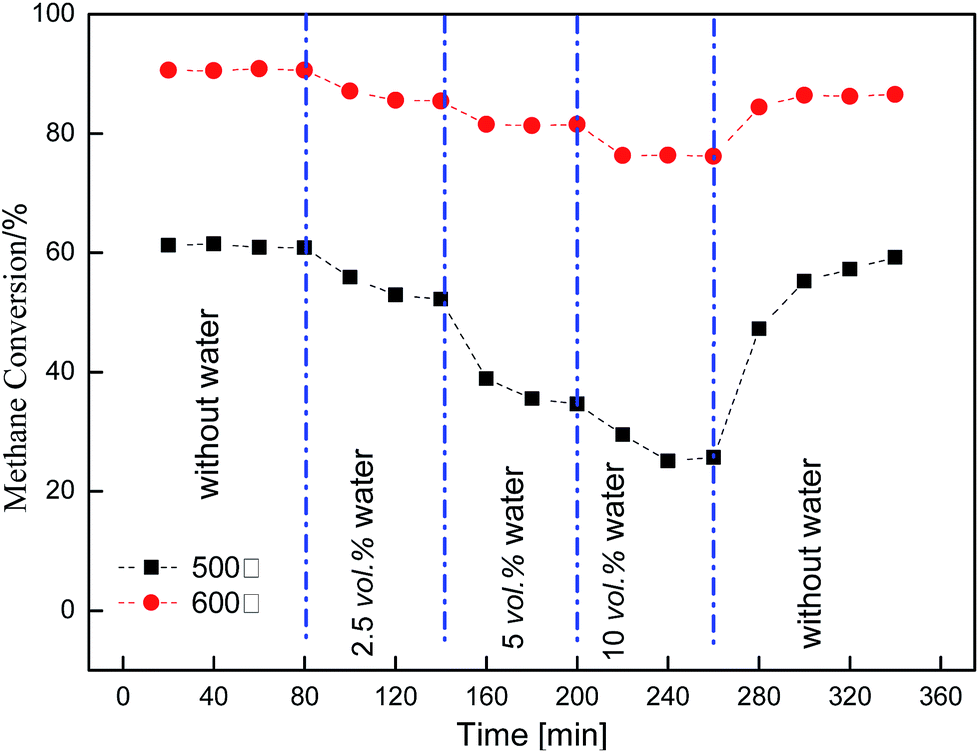 | ||
| Fig. 4 Water inhibitory effect on methane oxidation over the Cu/γ-Al2O3 catalyst (3 vol% CH4, 20 vol% O2). | ||
Different from Pd and Pt catalysts, the Cu catalyst displayed a stronger resistance to water and could maintain a stable activity at high steam pressure. For the inhibitory effect of water vapor, it was generally considered that water species could adsorb on the catalyst surface, and react with chemisorbed O (O*): H2O + O* → OH*. Thus, a portion of O* did not participate in methane oxidation, making methane conversion lower.9,12–14
From Fig. 4, if H2O flow was turned off, the activity almost returned to the initial level. This indicated there was some water adsorbed on the catalyst surface at low temperature. When the water was turned off, water pressure decreased. H2O gradually desorbed from the surface and O* was able to react with CH4.
3.2. Regeneration of activity of the catalyst
Regeneration of the catalytic activity of the Cu/γ-Al2O3 catalyst was studied by adding water cyclically to the feed gas. By controlling the reaction temperature (500 °C, 550 °C, 600 °C) and water concentration (2.5 vol%; 5 vol%; 10 vol%), Fig. 5 shows the effect when the catalyst was purged with N2. The feed stream was as follows: CH4, 3 vol%; O2, 20 vol%; H2O (2.5 vol%; 5 vol%; 10 vol%); N2, balance.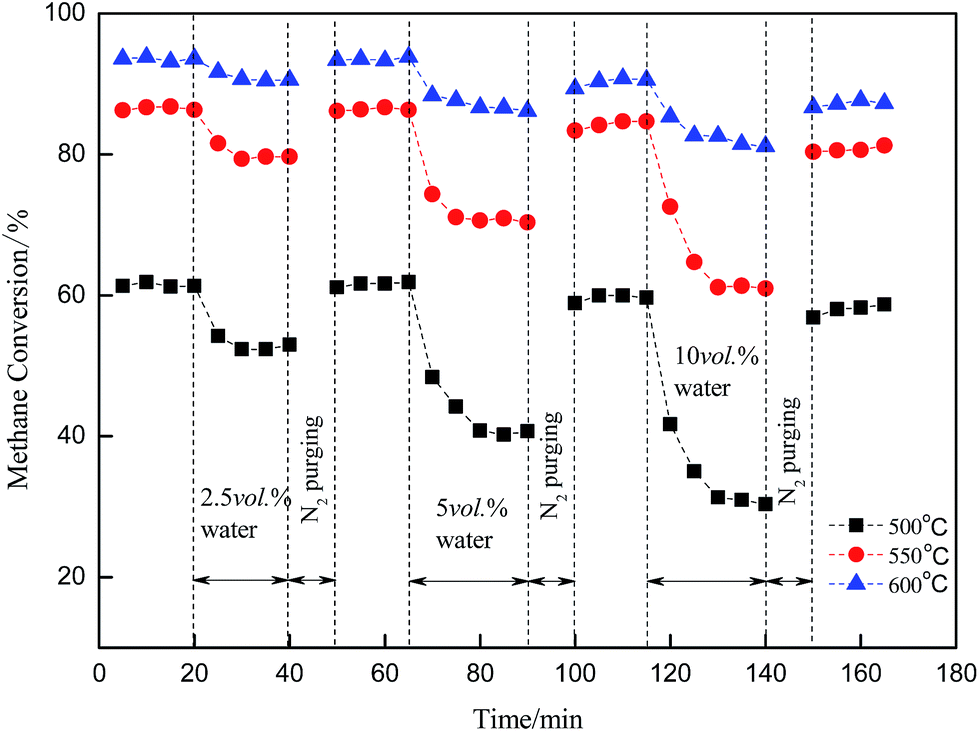 | ||
| Fig. 5 Regeneration of the catalytic activity of the Cu/γ-Al2O3 catalyst (3 vol% CH4, 20 vol% O2). | ||
In Fig. 5, CH4 conversion was measured by gas chromatography every 15 minutes, whereas the catalyst was purged with nitrogen in the absence of CH4 for another 10 minutes. Methane conversion was initially high. Then, the activity dropped as water was introduced into the feed gas. Thereafter, the catalyst was purged with N2 at the same temperature; consequently, no methane was shown in the figure. After purging, the activity of the catalyst almost recovered to its initial level. Comparisons of 500 °C, 550 °C and 600 °C show high temperature weakened the inhibitory effect and activity dropped by a smaller amount compared with low temperature. This meant H2O or surface OH* did not adsorb on the catalyst surface stably, and if temperature increased (500 °C → 600 °C), the process of desorption or decomposition was easy to proceed: OH* → H2O + O*.
3.3. Surface characteristics and stability
In order to discover the influence of water vapor on the catalyst surface, the SEM pictures (1000 times) for the catalyst surface are shown in Fig. 6 and BET surface areas (fresh catalyst or the catalyst after the reaction) are shown in Table 3. | ||
| Fig. 6 Surface characteristics of the Cu/γ-Al2O3 catalyst (SEM). (a) Fresh catalyst; (b) catalyst after reaction in the dry feed; (c) catalyst after reaction with 5 vol% water; (d) catalyst after reaction with 10 vol% water. | ||
| Without water | With water | ||
|---|---|---|---|
| Before reaction | After reaction | 5 vol% | 10 vol% |
| 188.2 (m2 g−1) | 180.2 (m2 g−1) | 166.8 (m2 g−1) | 152.6 (m2 g−1) |
In Table 3, the BET surface area of the fresh catalyst was 188.2 m2 g−1, while the catalyst after the reaction in the dry feed was 180.2 m2 g−1. The surface area was reduced by 8.0 m2 g−1 after the reaction. When 5 vol% or 10 vol% vapor was added into the feed, the BET surface area decreased to 166.8 m2 g−1 and 152.6 m2 g−1 respectively. Comparing Fig. 6(a) and (b) shows the sizes of catalyst surface grains were uniform after the reaction. However, in Fig. 6(c) and (d), when 5 vol% and 10 vol% water was introduced into the feed stream, the catalyst grains grew significantly and an agglomerate phenomenon obviously occurred.
According to sections 3.1 and 3.2, activity of the Cu/γ-Al2O3 catalyst did not recover to its initial level, in spite of high temperature or N2 purging. The reason might be that the morphology of particles was changing very quickly due to this heat and water treatment, and surface deformation blocked the active site. Additional water promoted the catalyst surface sintering and some active sites were covered or embedded into the support. Parts of the active sites did not interact with methane or oxygen, making the activity of the Cu/γ-Al2O3 catalyst decrease.
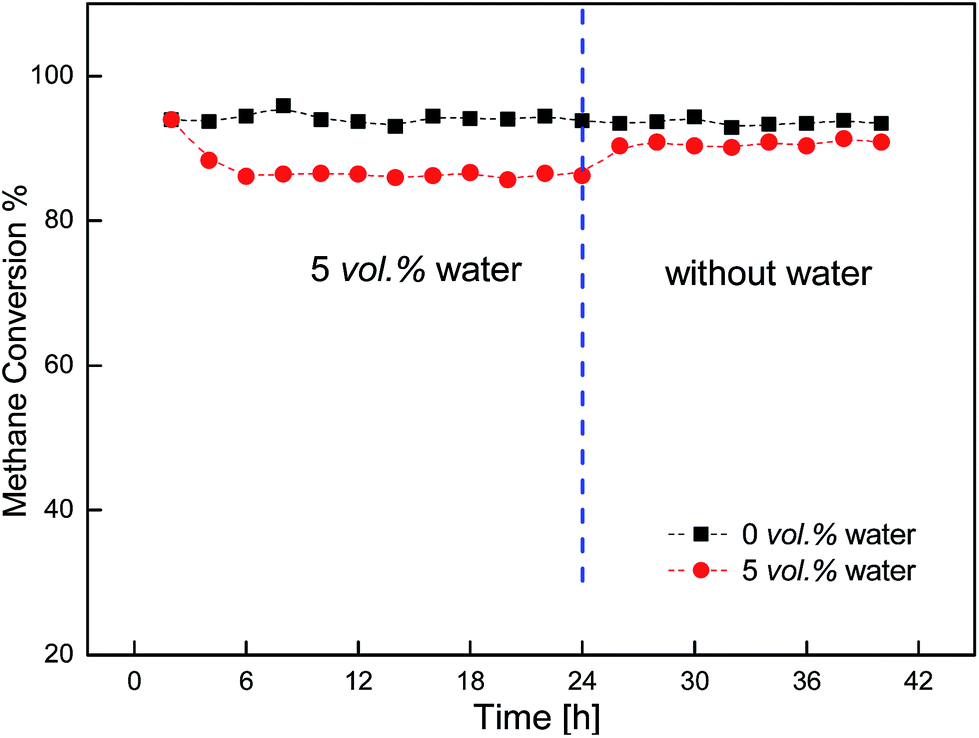
Fig. 7 showed the stability of the activity of the Cu/γ-Al2O3 catalyst for 42 h with and without water. During the 42 h, when 5 vol% water was introduced into the feed, the catalyst maintained high activity for a long time. As the water was turned off, reactivity could recover and maintain stable for 18 h.
 | ||
| Fig. 7 Stability of the activity of the Cu/γ-Al2O3 catalyst (600 °C). | ||
3.4. Apparent activation energy of methane
In order to get the apparent activation energy and kinetic parameters of methane combustion with water vapor, we controlled the reaction conditions and the catalyst to meet the differential conditions. Methane conversion is required to be limited in a low range (<10%) and the catalyst must be diluted to reduce the influence of mass transfer and temperature fluctuation.27,28 Therefore, we tested the intra-pellet dilution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, 1:15, 1:20, 1:25) and inter-pellet dilution (1:50, 1:75, 1:100) to obtain a suitable dilution ratio to reduce these influences. Fig. 8 shows the results of the dilution experiment. From Fig. 8, when the dilution ratios were 1:20 for intra-pellet dilution and 1:75 for inter-pellet dilution, the reaction rate reached stability. For intra-pellet dilution, 0.05 g Cu was chosen to load on 1 g γ-Al2O3, and this catalyst was mixed with 3.75 g γ-Al2O3 for inter-pellet dilution. The exact values of Cu loading on the support for intra-pellet dilution (1:20) were tested by XRF: Cu, 5.13%; Al, 43.64%; O, 45.63%; Si, 1.43%; K, 1.52%; other, 2.65%. Reaction rate r can be described as:
:10, 1:15, 1:20, 1:25) and inter-pellet dilution (1:50, 1:75, 1:100) to obtain a suitable dilution ratio to reduce these influences. Fig. 8 shows the results of the dilution experiment. From Fig. 8, when the dilution ratios were 1:20 for intra-pellet dilution and 1:75 for inter-pellet dilution, the reaction rate reached stability. For intra-pellet dilution, 0.05 g Cu was chosen to load on 1 g γ-Al2O3, and this catalyst was mixed with 3.75 g γ-Al2O3 for inter-pellet dilution. The exact values of Cu loading on the support for intra-pellet dilution (1:20) were tested by XRF: Cu, 5.13%; Al, 43.64%; O, 45.63%; Si, 1.43%; K, 1.52%; other, 2.65%. Reaction rate r can be described as:
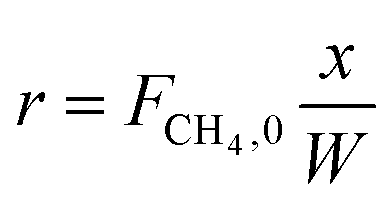 | (2) |
 | ||
| Fig. 8 Intra-pellet dilution and inter-pellet dilution of catalyst (1 vol% CH4, 400 °C). | ||
Temperature was controlled between 360–450 °C to limit methane conversion (<10%). The reaction conditions were: CH4, 3 vol%; O2, 20 vol%; H2O, 0–10 vol%; N2 balance. It is generally believed that, unlike the influence of O2 or CO2 on combustion in the oxygen-rich conditions, water has an obvious effect on activity. The reaction orders with respect to O2 and CO2 were zero and to methane was constant, nearly 1.
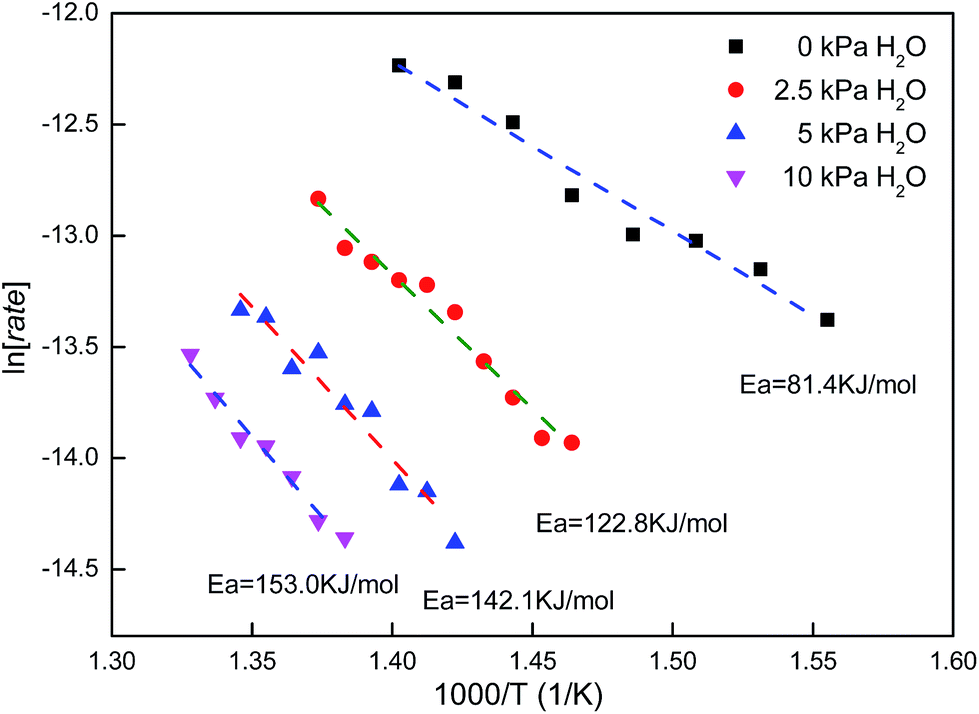
Fig. 9 shows the Arrhenius point linear fitting results of methane combustion with different water concentrations in the feed gas. As no water was introduced into the feed, apparent activation energy (Ea) was 81.4 kJ mol−1. When H2O was introduced into the feed (2.5 vol%, 5 vol%, 10 vol%), apparent activation energy (Ea) increased: 100.8 kJ mol−1, 113.8 kJ mol−1, 121 kJ mol−1. Therefore, water blocked methane oxidation.
 | ||
| Fig. 9 Arrhenius plots of reaction rate vs. 1/T for methane combustion (3 vol% CH4; 20 vol% O2; catalyst intra-pellet dilution, 1:20; catalyst inter-pellet dilution, 1:75). | ||
Using the method of Arrhenius point linear fitting, Van Giezen et al.22 also reported the apparent activation energy of methane over a Pd catalyst. They controlled the intake flow ratio: CH4, 1 vol%; O2, 4 vol%; N2 balance, H2O, 0 or 2 vol%, and assumed 0 order reactions with respect to oxygen and CO2, and constant reaction orders with respect to methane (1). Based on the assumptions above, they demonstrated the relationship between reaction order with respect to H2O and apparent activation energy:
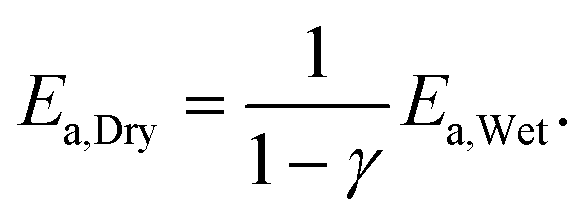 | (3) |
According to the above equation, we calculated the reaction order with respect to water: −0.6, −0.796 and −0.911, respectively. Reaction order with respect to water decreased as water concentration increased, but the value was within the scope of −0.6 to −1 and in agreement with the related reports.22,23 H2O influence mainly happened at low temperature (<550 °C), in which water adsorbed on O* easily. Apparent activation energy and reaction order with respect to water changed under certain conditions.
3.5. Water surface coverage
To explain the adsorption characteristics of vapor on the active site, we quote the Langmuir equation to describe water adsorption on the active site. We supposed that the adsorption of methane molecules on the active site was the rate-limiting step (oxygen-rich) and methane adsorption on the active site was dissociative and irreversible. Water molecules adsorbed onto the active site surface, and formed OH* groups. OH* could recombine to desorb as H2O molecules which returned to gaseous phase reactants, generating surface reduced sites. In this process, water and methane molecules were competing for adsorption on the active site. The rate equation can be expressed as:| r = k[CH4]θ | (4) |
Meanwhile, due to the lower partial pressure of methane as a gaseous reactant and rapid consumption with O2 after adsorption, methane surface coverage was much lower compared to water. Surface active vacancies could be expressed by means of the Langmuir isotherm:
 | (5) |
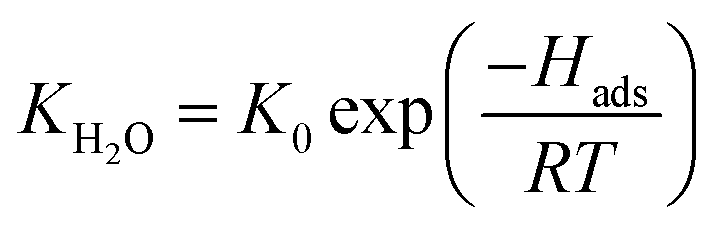 | (6) |
θH2O is the water surface coverage on the active site, KH2O and Hads are the water adsorption equilibrium constant and the water adsorption enthalpy, K0 the pre-exponential factor, and [H2O] the water concentration. When water was introduced into the feed, the expression is:
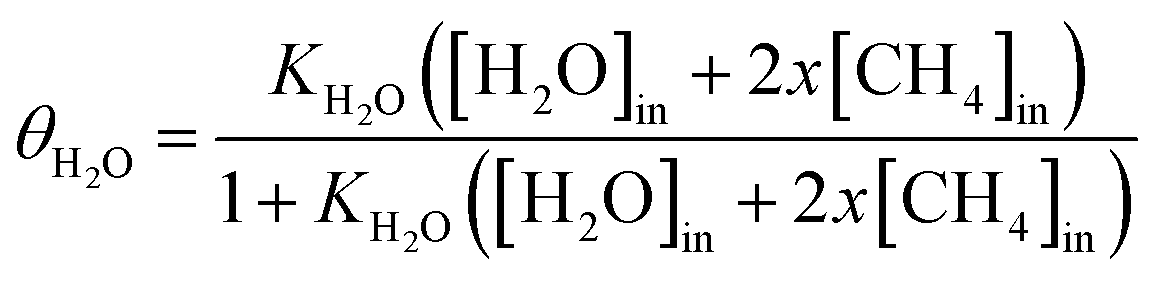 | (7) |
According to related reports and the results of density functional theory calculations,10,11 water adsorption enthalpy (Hads) over the Cu catalyst was about −120 to −160 kJ mol−1 and the pre-exponential factor (K0) was about a magnitude of 1010. Although this was a rough estimate, it obtained an acceptable value of θH2O as Hads and K0 were −160 kJ mol−1 and 1010, respectively. Calculating using eqn (7) with these parameters, we obtained water coverage with temperature.
Fig. 10 shows water coverage changing with temperature. Water concentrations were: 0 vol%, 2.5 vol%, 5 vol%,10 vol%. Without water in the feed gas, water coverage was below 30% at 400 °C. As water was introduced into the feed, water coverage rose above 87% at 400 °C. At each concentration (water: 2.5 vol%, 5 vol%, 10 vol%), θH2O had the same downtrend which tended towards 0% when the temperature rose up. At low temperature, there was an obvious difference between water coverage with or without water in the feed gas. But at high temperature, the coverage curve tended to the same. High temperature enhanced the desorption of water, making water coverage lower.
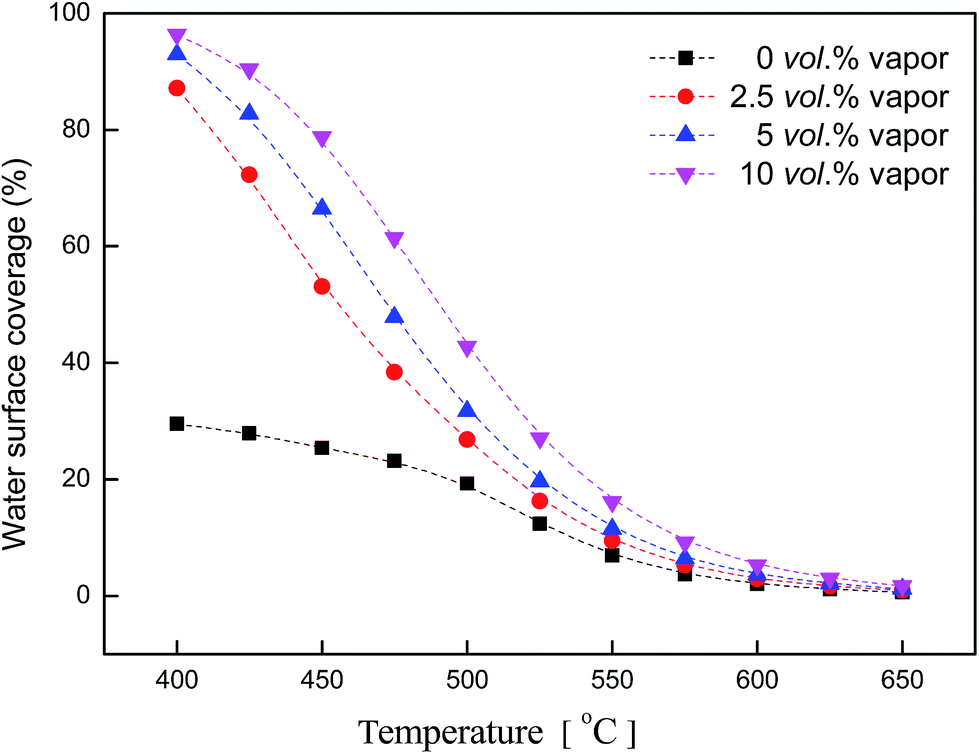 | ||
| Fig. 10 Water coverage vs. temperature (reaction conditions: CH4, 3 vol%; O2, 20 vol%; H2O, 0–10 vol%; N2 balance). | ||
4. Conclusion
The effect of water on methane combustion over the Cu/γ-Al2O3 catalyst was investigated in a fixed bed reactor. From the experimental results, we concluded the following points:1. Combustion efficiency of methane over the Cu/γ-Al2O3 catalyst was inhibited by H2O. The inhibitory effect was weakened with rising temperature. Because of reversible adsorption of water, reactivity could be recovered by N2 purging which promoted water molecule desorption from the active site.
2. Water vapor promoted the deformation of the catalyst surface, leading to the decrease of surface area and other irreversible effects.
3. The apparent activation energy of methane rose up noticeably as vapor was introduced into the feed and the reaction order with respect to water vapor was maintained between −0.6 and −1. Water surface coverage had a downtrend with temperature, which tended towards 0% above 625 °C.
Acknowledgements
The authors would like to thank the financial support of Natural Science Foundation of China with Project no. 51206200, and the Fundamental Research Funds for the Central Universities with Project no. CDJZR12140031, and visiting Scholar Foundation of Key Lab. of Low-grade Energy Utilization Technologies and System, MOE of China in Chongqing University (LLEUTS-201301).References
- T. Kai, Z. Shenghu and Z. Qiuju, et al., Sol–gel auto-combustion synthesis of Ni-CexZr1−xO2 catalysts for carbon dioxide reforming of methane, RSC Adv., 2013, 3(44), 22285–22294 RSC.
- Z. Yang, J. R. Grace, C. Jim Lim and L. Zhang, Combustion of low-concentration coal bed methane in a fluidized bed reactor, Energy Fuels, 2011, 25(3), 975–980 CrossRef CAS.
- Z. Yang, J. Liu and L. Zhang, et al., Catalytic combustion of low-concentration coal bed methane over CuO/γ-Al2O3 catalyst: effect of SO2, RSC Adv., 2014, 4(74), 39394–39399 RSC.
- B. Saurabh and V. Gtz, Chemical looping beyond combustion: Production of synthesis gas via chemical looping partial oxidation of methane, RSC Adv., 2014, 4(88), 47254–47267 RSC.
- G. Guangsheng, L. Kuo, W. Lijuan and G. Fubo, et al., High specific surface area LaMO3 (M = Co, Mn) hollow spheres: Synthesis, characterization and catalytic properties in methane combustion, RSC Adv., 2014, 4(102), 58699–58707 RSC.
- S. Eriksson, M. Boutonnet and S. Järås, Catalytic combustion of methane in steam and carbon dioxide-diluted reaction mixtures, Appl. Catal., A, 2006, 312(8), 95–101 CrossRef CAS PubMed.
- V. Dupont, S. H. Zhang and R. Bentley, et al., Experimental and modelling studies of the catalytic combustion of methane, Fuel, 2002, 81(6), 799–810 CrossRef CAS.
- H. Xun, Z. Lijun, D. Dehua and L. Gongxuan, High-temperature steam reforming of bio-oil derived light organics and methane to hydrogen-rich gas with trace CO via rational temperature control, RSC Adv., 2014, 4(36), 18924–18929 RSC.
- P. Gélin, L. Urfels and M. Primet, et al., Complete oxidation of methane at low temperature over Pt and Pd catalysts for the abatement of lean-burn natural gas fuelled vehicles emissions: influence of water and sulphur containing compounds, Catal. Today, 2003, 83(1), 45–57 CrossRef.
- K. Nagase, Y. Zheng and Y. Kodama, et al., Dynamic Study of the Oxidation State of Copper in the Course of Carbon Monoxide Oxidation over Powdered CuO and Cu2O, J. Catal., 1999, 187(1), 123–130 CrossRef CAS.
- M. Shinde Vijay and M. Giridhar, Catalytic performance of highly dispersed Ni/TiO2 for dry and steam reforming of methane, RSC Adv., 2014, 4(10), 4817–4826 RSC.
- D. Gao, S. Wang and C. Zhang, et al., Methane Combustion over Pd/Al2O3 Catalyst: Effects of Chlorine Ions and Water on Catalytic Activity, Chin. J. Catal., 2008, 29(12), 1221–1225 CrossRef CAS.
- R. Kikuchi, S. Maeda and K. Sasaki, et al., Low-temperature methane oxidation over oxide-supported Pd catalysts: inhibitory effect of water vapor, Appl. Catal., A, 2002, 232(1), 23–28 CrossRef CAS.
- P. Araya, S. Guerrero and J. Robertson, et al., Methane combustion over Pd/SiO2 catalysts with different degrees of hydrophobicity, Appl. Catal., A, 2005, 283(1), 225–233 CrossRef CAS PubMed.
- X. Hu, Q. Yu and N. Sun, et al., Experimental study of flammability limits of oxy-methane mixture and calculation based on thermal theory, Int. J. Hydrogen Energy, 2014, 39(17), 9527–9533 CrossRef CAS PubMed.
- D. Ciuparu, E. Perkins and L. Pfefferle, In situ DR-FTIR investigation of surface hydroxyls on γ-Al2O3 supported PdO catalysts during methane combustion, Appl. Catal., A, 2004, 263(2), 145–153 CrossRef CAS PubMed.
- D. Ciuparu and L. Pfefferle, Support and water effects on palladium based methane combustion catalysts, Appl. Catal., A, 2001, 209(1), 415–428 CrossRef CAS.
- B. Stasinska, A. Machocki and K. Antoniak, et al., Importance of palladium dispersion in Pd/Al2O3 catalysts for complete oxidation of humid low-methane–air mixtures, Catal. Today, 2008, 137(2), 329–334 CrossRef CAS PubMed.
- O. Wee-Jun, G. Meei Mei and C. Siang-Piao, et al. Direct growth of carbon nanotubes on Ni/TiO2 as next generation catalysts for photoreduction of CO2 to methane by water under visiblelight irradiation, RSC Adv., 2013, 3(14), 4505–4509 RSC.
- D. Qiao, G. Lu and Y. Guo, et al., Effect of water vapor on the CO and CH4 catalytic oxidation over CeO2–MOx (M= Cu, Mn, Fe, Co, and Ni) mixed oxide, J. Rare Earths, 2010, 28(5), 742–746 CrossRef CAS.
- W. Tao, C. Weiye and Z. Peng, et al., Cu-Ni@SiO2 alloy nanocomposites for methane dry reforming catalysis, RSC Adv., 2013, 3(46), 23976–23979 RSC.
- J. C. Van Giezen, F. R. Van den Berg and J. L. Kleinen, et al., The effect of water on the activity of supported palladium catalysts in the catalytic combustion of methane, Catal. Today, 1999, 47(1), 287–293 CrossRef CAS.
- G. Águila, F. Gracia and J. Cortés, et al., Effect of copper species and the presence of reaction products on the activity of methane oxidation on supported CuO catalysts, Appl. Catal., B, 2008, 77(3), 325–338 CrossRef PubMed.
- C. L. Pieck, C. R. Vera and E. M. Peirotti, et al., Effect of water vapor on the activity of Pt-Pd/Al2O3 catalysts for methane combustion, Appl. Catal., A, 2002, 226(1), 281–291 CrossRef CAS.
- Y. Ozawa, Y. Tochihara and M. Nagai, et al., PdO/Al2O3 in catalytic combustion of methane: stabilization and deactivation, Chem. Eng. Sci., 2003, 58(3), 671–677 CrossRef CAS.
- K. Persson, L. D. Pfefferle and W. Schwartz, et al., Stability of palladium-based catalysts during catalytic combustion of methane: The influence of water, Appl. Catal., B, 2007, 74(3), 242–250 CrossRef CAS PubMed.
- Y. H. Chin, C. Buda and M. Neurock, et al., Reactivity of chemisorbed oxygen atoms and their catalytic consequences during CH4–O2 catalysis on supported Pt clusters, J. Am. Chem. Soc., 2011, 133(40), 15958–15978 CrossRef CAS PubMed.
- J. Xu, L. Ouyang and W. Mao, et al., Operando and Kinetic Study of Low-Temperature, Lean-Burn Methane Combustion over a Pd/γ-Al2O3 Catalyst, ACS Catal., 2012, 2(2), 261–269 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |