
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel photolabile crosslinkers based on O-acyloxime moiety†
Kanji Suyama *a and
Hideki Tachib
*a and
Hideki Tachib
aFaculty of Liberal Arts and Sciences, Osaka Prefecture University, 1-1, Gakuen-Cho, Naka-Ku, Sakai, Osaka 599-8531, Japan. E-mail: suyama@las.osakafu-u.ac.jp
bTextile & Polymer Section, Technology Research Institute of Osaka Prefecture, 7-1 Ayumino-2, Izumi, Osaka 594-1157, Japan
First published on 25th March 2015
Abstract
Controlled decrosslinking is an attractive technique in the fields of functional materials and polymer recycling. Photo-triggered decrosslinking is especially advantageous for easy remote activation and spatiotemporal control. However, only a few photolabile units have been used for the cleavage in crosslinkers. Herein, 1,4-diacetylbenzene 1,4-bis(O-methacryloyl)dioxime and 1,3,5-triacetylbenzene 1,3,5-tris(O-methacryloyl)trioxime are proposed as novel crosslinkers. They have a benzene ring bearing two or three arms in which O-acyloxime moiety is introduced as a novel type of photolabile unit. These crosslinkers were prepared from aromatic ketones in two steps. Photopolymerization of methyl acrylate and the crosslinkers was performed using diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide as a photoinitiator and UV light above 310 nm. Obtained polymer films were crosslinked and insoluble in tetrahydrofuran (THF). These films were further irradiated at 254 nm of light. The photolysis of O-acyloxime units in these crosslinkers were confirmed by UV and IR spectral measurements. The precedence of decrosslinking on 254 nm irradiation was evidenced by the decrease in thickness of the films after soaking in THF. Soluble fraction of the decrosslinked polymers were subjected to size exclusion chromatography, and their molecular weights were found to be similar to that obtained by photopolymerized film in the absence of the crosslinker. These results demonstrated that the proposed compounds are potential candidates for novel photolabile crosslinkers.
Introduction
Crosslinked polymers show excellent mechanical strength as well as thermal and chemical stability compared to linear polymers. Therefore, decrosslinking can give rise to drastic changes in many properties of the crosslinked polymers. In this regard, the incorporation of the units that leads to the decrosslinking on demand is an attractive design for functional materials. Furthermore, decrosslinking technology provides an important process in the field of polymer recycling.1Several stimuli such as heat,2–6 pressure,7 electrical voltage,8 ultrasonic,9 the addition of acids,10–12 bases,3,12–14 and other reagents,15,16 and light3,7,17–34 have been applied to trigger the decrosslinking. Among them, photochemical process is appealing because of its easy remote activation and spatiotemporal control. These features have been applied in the fields of nanoimprint lithography,17,18 drug delivery,19 and self-healing materials.20
Photo-triggered decrosslinking can be accomplished by incorporating photolabile units into main-chain or linker units. Although indirect methods which utilize photo-triggered release of catalysts such as strong acids have been proposed recently,17,18,21–25 the most versatile approach is to design crosslinkers bearing both polymerizing and photolabile units and to polymerize them. However, in most cases, the photolabile units used in the crosslinkers were limited to o-nitrobenzyl-26–32 and coumarine-based33,34 families. Especially, crosslinkers bearing three or higher polymerizing groups are rare.33,34
Recently, we have proposed 1,4-diacetylbenzene 1,4-bis(O-methacryloyl)dioxime (DBzM) and 1,3,5-triacetylbenzene 1,3,5-tris(O-methacryloyl)trioxime (TBzM) as novel photolabile crosslinkers.35 The chemical structures of these crosslinkers have O-acyloxime and methacryl units as photolabile and polymerizing units, respectively, as shown in Fig. 1.
 | ||
| Fig. 1 O-Acyloxime-based crosslinkers. | ||
These crosslinkers could be derived from starting ketones in two steps. The short synthetic route is advantageous to easy preparation of multifunctional monomers. Also, O-acyloxime moieties show good photo-reactivity and thermal stability. For example, quantum yield of photolysis at 254 nm and onset thermal decomposition temperature of 1,3,5-triacetylbenzene 1,3,5-tris(phenylacetyl)trioxime were 0.41 and 208 °C, respectively.36
As polymerizing units, we used methacrylates in this study. It has already found that methacrylates can serve six-membered transition state due to the presence of β-hydrogen, and finally turned into vinylidene double bond on main-chains and an imine as shown in Scheme 1(i).37,38 In case of acrylates, the coupling of C radical and imino radical is a major reaction to form a ketimine which is hydrolyzed into an amine and a ketone as Scheme 1(ii). We have been studied the photochemical changes of various O-acyloximes in polymer matrixes and found that the hydrolysis of ketimine proceeded immediately in polymer films with 0.3–0.4 μm thickness.36,39,40 Because we used thicker films in this study, we avoided acrylates that require the hydrolysis process for complete cleavage.
 | ||
| Scheme 1 Photoreactions of pendant O-acyloxime moieties. | ||
In this article, we report the photochemical behaviors of DBzM and TBzM in detail, especially comparing with that of polymers without the crosslinkers. We also analyzed the photodegradation process with spectroscopic and molecular weight measurements.
Experimental
Instruments
Simultaneous thermogravimetry-differential thermal analysis (TG-DTA) measurements were carried out using a Seiko Instruments TG/DTA320 with a heating rate of 10 K min−1 under N2. Mass spectra were taken with a Thermo Scientific Q Exactive by using positive electrospray ionization (ESI†).NMR, IR, and UV spectra were recorded by Jeol JNM-ECX400, Jasco FT-IR4200, and Jasco V530 spectrometers, respectively. Elemental analyses were carried out by a Perkin Elmer 2400 Elemental Analyzer.
Number (Mn) and weight (Mw) average molecular weights of polymers were obtained by size exclusion chromatography (SEC) using a system composed of Tosoh two sequential TSKgel HXL columns, a Tosoh DP8020 pump, and a Viscotek TriSEC 302W detector with THF eluent at 40 °C. In this experiment, the molecular weights were estimated using refractometer detector calibrated with polystyrene standards. Sample solutions were filtered with 0.2 μm pore sized PTFE disposable filters.
The formulations for photopolymerization were treated with a Shibata glass digifit dispenser (Tokyo, Japan). Thickness of films on substrates was measured by a Mitsutoyo SJ210 contact profilometer (Kawasaki, Japan) using a 5 μmϕ tip stylus with 4 mN measuring force.
Heating was performed on a Koike Precision Instruments KPI HP-19U300 hot plate (Itami, Hyogo, Japan).
Light sources for photopolymerization and photodegradation were performed with a Hayashi Watch-Works LA410 Xe–Hg lamp (Tokyo, Japan) and an Ushio ULO-6DQ low pressure lamp, respectively. Their light intensities were measured by an Orc UV-M03 illuminometer (Tokyo, Japan).
Materials
Methyl acrylate (MA) was purchased from Nacalai Tesque (Kyoto, Japan) and filtered with activated neutral alumina (Aldrich) to remove polymerization inhibitors. Diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) (Tokyo Chemical Industry) was recrystallized from hexane. Tetrahydrofuran (THF) was distilled before use. 1,3,5-Triacetylbenzene trioxime was prepared as described in literature.41Poly(ethylene terephthalate) (PET) films for covering the formulation was Toyobo Lumilar whose thickness was 50 μm. The transmittance at 313 and 365 nm were and 7 and 79%, respectively. Polyethylene (PE) films with 80 μm thick were used for the measurement of IR changes.
1,4-Diacetylbenzene dioxime
In a flask, 16.2 g (10.0 mmol) of 1,4-diacetylbenzene, 16.4 g (236 mmol) of hydroxylamine hydrochloride, 70 mL of methanol, and 10 mL of water were refluxed for 1 h. After cooling, the mixture was filtered to afford 17.6 g of ivory fine powder. From the filtrate, 2.33 g of powder was recovered when concentrated to ca. 30 mL. IR spectra of both powders showed a small C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band at 1680 cm−1. Thus, both powders were put together in a flask with 70 mL of methanol and 3.0 mL of hydroxylamine solution (Wako, 50%) and refluxed for 30 min. After cooling and filtration, recovered powder was refluxed for 1 h with 60 mL of ethanol and 3.0 mL of the hydroxylamine solution. Finally, 17.6 g (91.5 mmol) of colorless fine powder was obtained. Yield: 91.5% 1H NMR (DMSO-d6): 2.14 (6H, s, CH3), 7.62 (4H, s, aromatic), 11.24 (2H, s, OH). 13C NMR (DMSO-d6): 11.93, 126.02, 137.53, 153.15. IR (KBr): 2600–3500 cm−1 (OH).
O stretching band at 1680 cm−1. Thus, both powders were put together in a flask with 70 mL of methanol and 3.0 mL of hydroxylamine solution (Wako, 50%) and refluxed for 30 min. After cooling and filtration, recovered powder was refluxed for 1 h with 60 mL of ethanol and 3.0 mL of the hydroxylamine solution. Finally, 17.6 g (91.5 mmol) of colorless fine powder was obtained. Yield: 91.5% 1H NMR (DMSO-d6): 2.14 (6H, s, CH3), 7.62 (4H, s, aromatic), 11.24 (2H, s, OH). 13C NMR (DMSO-d6): 11.93, 126.02, 137.53, 153.15. IR (KBr): 2600–3500 cm−1 (OH).
DBzM
In a 200 mL of two-necked flask, 5.77 g (30.0 mmol) of 1,4-diacetylbenzene dioxime and 15.5 mL (112 mmol) of triethylamine were dissolved in 150 mL of dry dichloromethane, and a thermometer and a dropping funnel containing 10.0 mL (10.8 g, 103 mmol) of methacryloyl chloride were equipped. The flask was cooled in a bath at −20 °C, and the methacryloyl chloride was added dropwise over 8 min with stirring. The internal temperature was below −15 °C. Then the cooling bath was removed, and the mixture was stirred for 24 h at ambient temperature. The mixture was washed with 1.2 N HCl and sat. NaHCO3, and dried over MgSO4 for 2 h. After removing the solvent from the solution, 10.96 g of yellowish solid was obtained. The solid was recrystallized twice (from toluene–hexane and from methanol) to afford 6.34 g (19.3 mmol) of target monomer as ivory powder. Yield: 64.3% mp: 156 °C (DTA onset). 1H NMR (CDCl3): 2.07 (6H, s, CH3–CNO–), 2.44 (6H, s, methacryl CH3), 5.70 (2H, s, CH), 6.25 (2H, s, CH), 7.84 (4H, s, aromatic). 13C NMR (CDCl3): 14.39, 18.40, 126.45, 127.23, 135.22, 136.78, 162.46, 164.32. IR (KBr): 1738 (CO), 1634, 1606, 1317, 1122 (C–O–C) cm−1. UV: λmax (CH3CN) 275 nm (ε 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 dm3 mol−1 cm−1). ε at 254 nm was 16300 dm3 mol−1 cm−1. Elemental analysis, found: C, 65.55; H, 6.16; N, 8.76. Calc. for C18H20N2O4: C, 65.84; H, 6.14; N, 8.53%. MS/MS (ESI): m/z [M + Na]+ 351.
600 dm3 mol−1 cm−1). ε at 254 nm was 16300 dm3 mol−1 cm−1. Elemental analysis, found: C, 65.55; H, 6.16; N, 8.76. Calc. for C18H20N2O4: C, 65.84; H, 6.14; N, 8.53%. MS/MS (ESI): m/z [M + Na]+ 351.
TBzM
In a 200 mL of two-necked flask, 4.43 g (17.8 mmol) of 1,3,5-triacetylbenzene trioxime and 24 mL (172 mmol) of triethylamine were dissolved in 150 mL of dry dichloromethane, and a thermometer, and a dropping funnel containing 15.0 mL (16.2 g, 155 mmol) of methacryloyl chloride were equipped. After the flask was cooled in a bath at −30 °C, the methacryloyl chloride was added dropwise over 10 min with stirring. The internal temperature was below −10 °C. Then the cooling bath was removed, and the mixture was stirred for 24 h at ambient temperature. The solution was quenched with methanol with cooling in an ice bath. Then, the mixture was washed with 1.2 N HCl and sat. NaHCO3, and dried over Na2SO4 overnight. After the drying, the solution contained gelatinous materials which were removed by filtering. Resulting solution was rotavapped to afford 19.6 g of amber tar. The tar was washed with hexane 3 times, and recrystallized from methanol. Finally, obtained solid was column chromatographed with dichloromethane:ethyl acetate = 5:1 (v/v) to afford 2.25 g (4.96 mmol) of target monomer as colorless powder. Yield: 27.9% mp: 178 °C (DTA onset). 1H NMR (CDCl3): 2.09 (9H, s, CH3–CNO–), 2.50 (9H, s, methacryl CH3), 5.73 (3H, s, CH), 6.27 (3H, s, CH), 8.27 (3H, s, aromatic). 13C NMR (CDCl3): 14.73, 18.46, 126.66, 127.72, 135.19, 135.96, 162.44, 164.35. IR (KBr): 1747 (CO), 1635, 1621, 1373, 1317, 1289, 1279, 1139 (C–O–C), 1119 (C–O–C) cm−1. UV: λmax (CH3CN) 245 nm (ε 49200 dm3 mol−1 cm−1). ε at 254 nm was 42100 dm3 mol−1 cm−1. Elemental analysis, found: C, 63.44; H, 5.87; N, 9.39. Calcd for C24H27N3O6: C, 63.56; H, 6.00; N, 9.27%. MS/MS (ESI): m/z [M + Na]+ 476.
Photopolymerization
Formulations composed of DBzM:TPO:MA and TBzM:TPO:MA = 1.5:1:1000 and 1:1:1000 (mol/mol), respectively, were prepared for photopolymerization. After bubbling with nitrogen gas, 20 μL of the formulation was put on a quartz disc (20 mmϕ) with the dispenser, covered with a PET film (20 mmϕ), and irradiated with the Hg–Xe lamp for 60 s. The light intensity at 365 nm was 250 mW cm−2.
For the measurement of IR changes, CaF2 discs (20 mmϕ, 2 mm thick) were used as substrates. In this case, we covered the formulation with PE and PET films, and irradiated for 120 s with the Hg–Xe lamp, because polymerized films often remained on PET films rather than CaF2 discs. All irradiated films were baked at 80 °C for 2 min on a hot plate after removing the covering films.
The insoluble faction of photopolymerized films was calculated by measuring the weight of 4 sample films before and after soaking in THF for 3 min followed by baking at 80 °C for 2 min.
Photodegradation
Photodegradation of crosslinked polymers was carried out on irradiation at 254 nm with intensity of 1.8 mW cm−2. Irradiated films were soaked in THF for 3 min at room temperature followed by baking at 80 °C for 2 min. The films were scratched with a needle, and resulting cross-sections were measured with the surface roughness tester before the soaking. After soaking, the thickness was measured again. Normalized film thickness was calculated from the ratio of thickness before and after the soaking.Foe SEC measurement, decrosslinked polymers were recovered by rotavap of the THF solutions.
Results and discussion
Synthesis and properties of crosslinkers
Starting from corresponding aromatic ketones, DBzM and TBzM were obtained in two steps. The yields of the latter was relatively low because of the proceeding of polymerization during working up. Solubility of crosslinkers in organic solvents was not so high. For example, the solubility of DBzM and TBzM in MA was ca. 0.57 and 0.55 wt% at room temperature.TG-DTA profiles of DBzM and TBzM indicate small endoenergetic peaks around 160 and 180 °C, respectively, which were followed by exothermic peaks (ESI Fig. S5†). No DTA peak was observed below their melting points, suggesting that the thermal decomposition did not occur below these temperatures. The observed thermal stability fulfills the demand often required for imaging systems.42
The copolymerization of monofunctional O-methacryloyl oximes with common monomers such as styrene38 and methyl methacrylate43 was already published. Thus DBzM and TBzM can be expected to copolymerize with a variety of vinyl monomers.44
In UV spectra in acetonitrile, absorption maximum due to π–π* transition of DBzM appears at 275 nm, although that in TBzM is at 245 nm as shown in Fig. 2. Similar bathochromic shift of para-disubstituted benzenes compared to 1,3,5-trisubstituted ones is also found in starting ketones as well as other substituents such as cyano45 and alkyl groups,46 probably due to the participation of imino moieties in the resonance with benzene rings.46
 | ||
| Fig. 2 UV spectra of 1.0 × 10−5 M acetonitrile solutions of DBzM, TBzM, and TPO. Inset shows UV spectra of 1.2 × 10−3 M acetonitrile solution of TPO. | ||
Fig. 3a shows IR spectra of DBzM in KBr, where peaks due to CO and C–O stretching bands in O-acyloxime moiety appeared at 1738 and 1122 cm−1, respectively. When the KBr pellet was irradiated at 254 nm, these peaks decreased and a new peak appeared at 1684 cm−1. These characteristic peaks and their photochemical changes were also observed for TBzM as shown in Fig. 3b, where peaks due to CO and C–O stretching bands in O-acyloxime moiety at 1747, 1139, and 1119 cm−1 decreased on irradiation at 254 nm, and new peak appeared at 1688 cm−1. These IR spectral changes suggested the proceeding of photolysis of O-acyloxime moieties in Scheme 1(i). In aromatic imines, CN stretching bands appear in the region of 1620–1635 cm−1 with medium intensity.47 Therefore, the new peaks for DBzM and TBzM at 1684 and 1688 cm−1, respectively, are assigned to aromatic ketones rather than imines. Considering the moisture in KBr pellet, generated imine units would be hydrolyzed immediately.
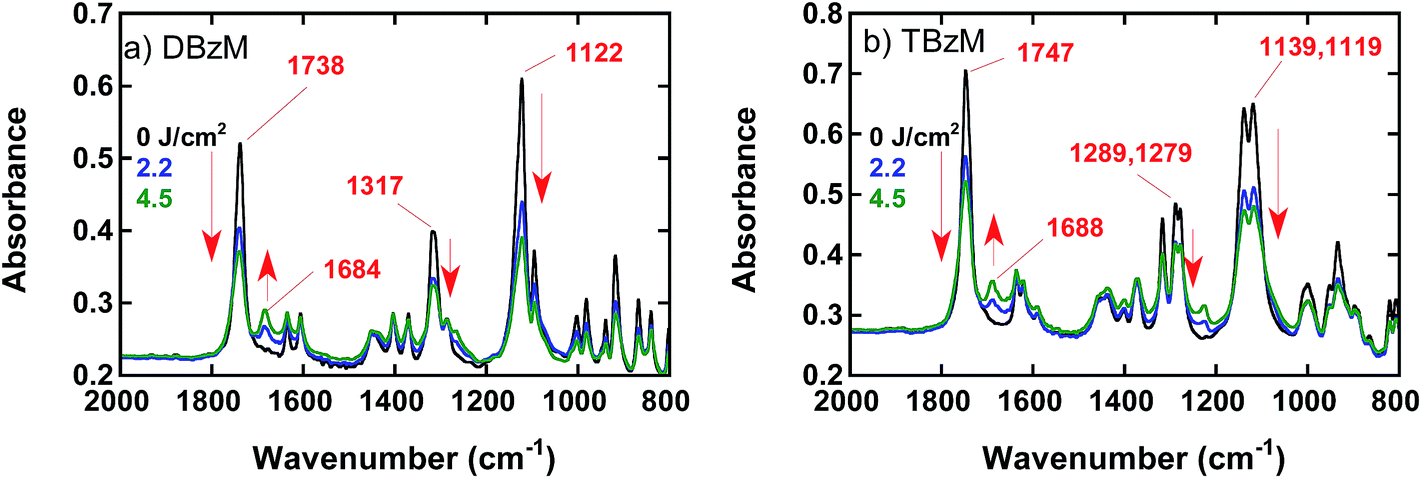 | ||
| Fig. 3 IR spectral changes of (a) DBzM and (b) TBzM in KBr on irradiation at 254 nm. Numbers in the figure show irradiation energy and wavenumbers of peak tops. | ||
Photopolymerization
Photopolymerization of MA in the absence and presence of crosslinkers was performed with TPO as a photoinitiator and a light above 310 nm. In the absence of crosslinker, resulting homopolymer (pMA) was not hard film but viscous tar.48 However, SEC profile revealed that Mn and Mw of pMA were 57600 and 324000, respectively. Both the greater value of polydispersity and a shoulder at higher molecular weight region in SEC profile (Fig. S6†) suggest that inhomogeneous proceeding of polymerization occurred in bulk polymerization.
When 0.15 mol% of DBzM against MA were involved in the formulation, obtained copolymer (DBzM–MA) gave films. Similarly, MA containing 0.10 mol% of TBzM turned into a copolymer (TBzM–MA) films. These films were hard and smooth-surfaced with 5 μm thickness. In addition, the insoluble fractions of DBzM–MA and TBzM–MA after soaking in THF were 52 and 79 wt%, respectively. These results show the formation of networked structure containing photolabile crosslinkers.
During the photopolymerization, the proceeding of simultaneous photodegradation could be considered. The photolysis of O-acyloxime moiety by using Hg–Xe lamp is possible in two ways: one is direct photolysis with 313 nm light, and the other is sensitized one by TPO with 365 nm light, because TPO involves benzoyl moiety which are known to sensitize the photoreaction of O-acyloximes as triplet sensitizers.49,50 However, judging from the insolubility of the polymers in THF, the degradation of O-acyloxime units was negligible. It is suggested that the direct photolysis was reduced by the low transmittance of the PET film at 313 nm (7%) and lower molar absorption coefficients of crosslinkers at 313 nm compared to those at 254 nm (DBzM: 7.5%, TBzM: 0.9%), and the sensitized photolysis did not occur due to low concentration of TPO and crosslinkers in solid films.
Photodegradation
The polymer films were further irradiated at 254 nm. Fig. 4 shows the UV spectral changes of these films. In the absence of crosslinkers, pMA film had a small absorbance at 254 nm, and there was little change in their spectra as shown in Fig. 4a. In the presence of crosslinkers, a peak at 274 nm in DBzM–MA (Fig. 4b) and a shoulder around 250 nm in TBzM–MA films (Fig. 4c), both are assigned to the π–π* transition of benzene units bearing O-acyloximes, decreased on irradiation at 254 nm. Although simple first-ordered decay was not observed in case of DBzM–MA as shown in Fig. 4d, the decrease for both polymers suggests that the photoreaction of O-acyloximes proceeded on irradiation at 254 nm.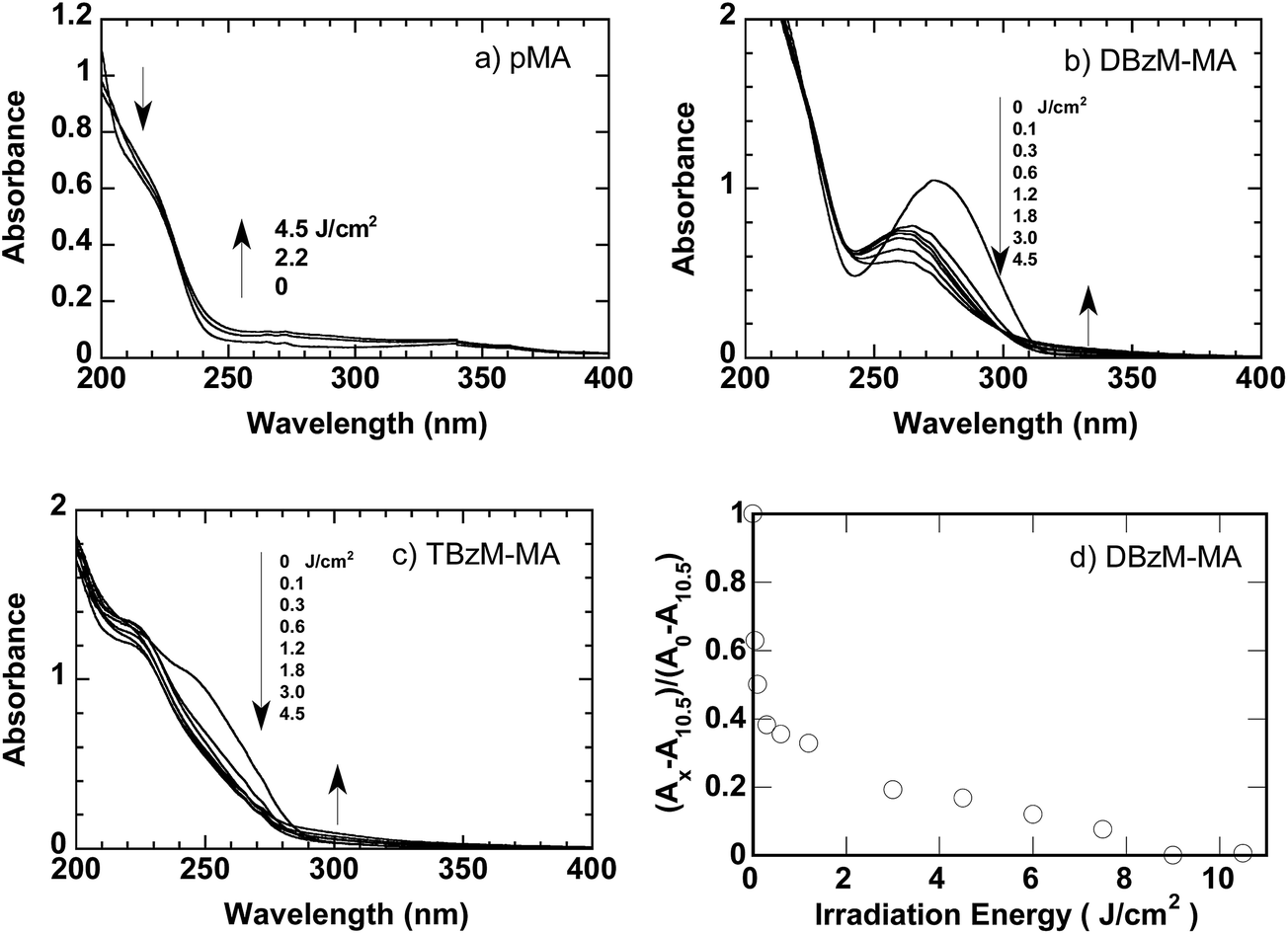 | ||
| Fig. 4 UV spectral changes of (a) pMA, (b) DBzM–MA, and (c) TBzM–MA films on irradiation at 254 nm. Numbers in the figures show irradiation energy. Film thickness: 5 μm. (d) Changes of normalized absorbance of DBzM–MA film as a function of irradiated energy at 254 nm. Ax indicates the absorbance at 274 nm with irradiation energy of x J cm−2. | ||
The changes of IR spectra of polymerized films were also measured. Although the molar feed of the crosslinkers was low, the changes of peaks due to the crosslinkers could be detected in difference spectra between non-irradiated and irradiated films as shown in Fig. 5.
 | ||
| Fig. 5 Changes of difference IR spectra between non-irradiated and 254 nm irradiated films of (a) pMA, (b) DBzM–MA, and (c) TBzM–MA. Numbers in the figure show irradiation energy at 254 nm and wavenumbers of peak tops. (d) Changes of normalized absorbance of TBzM–MA film as a function of irradiated energy at 254 nm. Ax indicates the absorbance at 1764 cm−1 with irradiation energy of x J cm−2. | ||
In Fig. 5a, the decrease at 1736 and 1161 cm−1 and increase at 1712 cm−1 were observed in pMA films. The reason of these changes is unclear, although it might be assigned to the slight shift of strong ester peaks in MA on irradiation at 254 nm. Fig. 5b showed the changes of difference spectra for DBzM–MA films. Unfortunately, the changes were similar to those with pMA, because the characteristic peaks due to CO and C–O stretching bands in O-acyloxime unit overlapped with those in MA. However, DBzM–MA film showed a small increase in the region of 1620–1712 cm−1. This increase suggested that imine units were produced as shown in Scheme 1(i), and a part of the imine units were hydrolyzed to ketone units. In case of TBzM–MA, the decrease at 1764 cm−1 due to CO stretching band in O-acyloxime unit was clearly observed in Fig. 5c, and the decrease was monotonous as plotted in Fig. 5d. In addition, the appearance of peaks at 1688 and 1645 cm−1 were observed, which are assignable to CO stretching band in aromatic ketones and CN stretching band in aromatic imines, respectively. From the above spectral changes, the plausible photochemical reactions of crosslinkers in polymers are summarized in Scheme 2.
 | ||
| Scheme 2 Plausible photoreaction of O-acyloxime-based crosslinkers. | ||
Photopolymerized films with different irradiation energies were soaked in THF. Although both DBzM–MA and TBzM–MA films were almost insoluble without irradiation at 254 nm, film thickness decreased along with an increase in irradiation energy at 254 nm irradiation as shown in Fig. 6. These results clearly indicate the proceeding of decrosslinking to form linear polymer chains as shown in Scheme 2. The remaining thickness for TBzM–MA films was slightly higher than those of DBzM–MA films. This may be due to the higher functionality of the crosslinker to maintain networked structure.
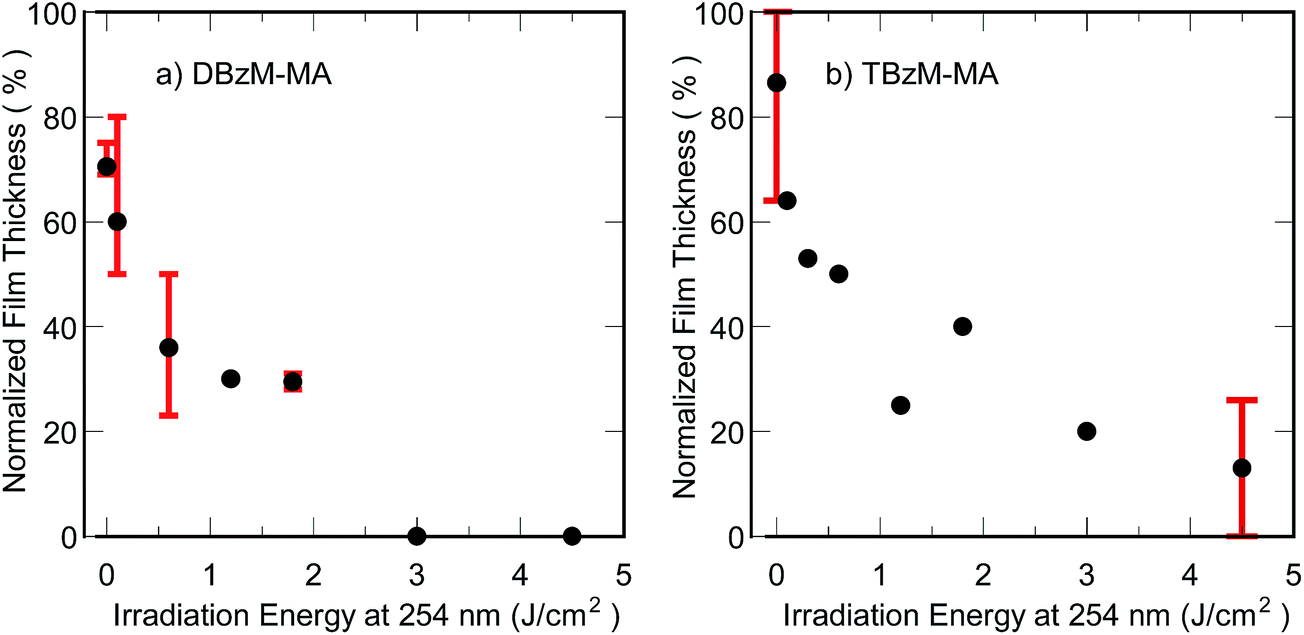 | ||
| Fig. 6 Thickness changes of (a) DBzM–MA and (b) TBzM–MA films after soaking in THF as a function of irradiation energy at 254 nm. Film thickness before soaking: 5 μm. | ||
Soluble fractions in THF were collected, filtered, and their molecular weights were measured by SEC. Table 1 summarizes the molecular weights, where Mn of TBzM–MA was slightly smaller than that of DBzM–MA. This tendency is consistent with the discussion on the relationship between molecular weight and crosslinker functionality,51,52 where trifunctional crosslinkers tended to form more rigid networks and thus to give lower molecular weight between crosslinking points compared to difunctional ones. However, the SEC profiles of pMA, DBzM–MA, and TBzM–MA were almost identical as observed in Fig. S6.† In order to discuss the effect of the functionality, other experimental condition such as higher concentration of the crosslinkers might be needed. The small decrease in molecular weights for pMA on irradiation at 4.5 J cm−2 would be due to proceeding of main-chain scission and volatile products formation, which has often been found on irradiation in the presence of oxygen.53
Conclusion
Novel photolabile crosslinkers, DBzM and TBzM were prepared and characterized. Both crosslinkers were successfully photopolymerized with MA on irradiation at >310 nm in the presence of TPO. The photolysis of O-acyloxime units in the resulting polymers at 254 nm was confirmed by UV and IR difference spectral changes, which led to the degradation of the networked structure. SEC analysis revealed that the molecular weights of linear pMA were similar to those of DBzM–MA and TBzM–MA after irradiation at 254 nm. These results demonstrated that DBzM and TBzM are potential candidates for novel photolabile crosslinkers.Acknowledgements
The authors thank to Dr Hirokazu Hayashi for measuring MS spectra of crosslinkers. This work was supported by JSPS KAKENHI Grant number 25340095.References and notes
- H. Nishida, Polym. J., 2011, 43, 435 CrossRef CAS.
- K. Ogino, J.-S. Chen and C. K. Ober, Chem. Mater., 1998, 10, 3833 CrossRef CAS.
- S. Taketani and A. Matsumoto, Macromol. Chem. Phys., 2004, 205, 2451 CrossRef CAS.
- Z. Chen, L. Zhao and Z. Wang, Polymer, 2013, 54, 5182 CrossRef CAS PubMed.
- H. Otsuka, Polym. J., 2013, 45, 879 CrossRef CAS.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1 CrossRef CAS PubMed.
- T. Iwamura and S. Nakamura, Polymer, 2013, 54, 4161 CrossRef CAS PubMed.
- T.-W. Chuo, T.-C. Wei and Y.-L. Liu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3395 CrossRef CAS.
- K. Huang, A. I. Isayev and J. Zhong, J. Appl. Polym. Sci., 2014, 131, 40680 Search PubMed.
- R. R. De Clercq and E. J. Goethals, Macromolecules, 1992, 25, 1109 CrossRef CAS.
- T. Hashimoto, H. Meiji, M. Urushisaki, T. Sakaguchi, K. Kawabe, C. Tsuchida and K. Kondo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3674 CrossRef CAS.
- Y. Kohsaka, K. Nakazono, Y. Koyama, S. Asai and T. Takata, Angew. Chem., Int. Ed., 2011, 50, 4872 CrossRef CAS PubMed.
- G. K. Paul and S. Ramakrishnan, Macromol. Chem. Phys., 2001, 202, 2872 CrossRef CAS.
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, Macromolecules, 2014, 47, 498 CrossRef CAS.
- X. Hu, H. Li, S. Luo, T. Liu, Y. Jiang and S. Liu, Polym. Chem., 2013, 4, 695 RSC.
- M. Yonekawa, Y. Furusho, T. Takata and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 921 CrossRef CAS.
- M. Shirai, Prog. Org. Coat., 2007, 58, 158 CrossRef CAS PubMed.
- M. Shirai, Polym. J., 2014, 46, 859 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS PubMed.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446 RSC.
- H. Okamura and M. Shirai, Polymer, 2010, 51, 5087 CrossRef CAS PubMed.
- G. Huh, K.-O. Kwon, S.-H. Cha, S.-W. Yoon, M. Y. Lee and J.-C. Lee, J. Appl. Polym. Sci., 2009, 114, 2093 CrossRef CAS.
- D. Matsukawa, H. Okamura and M. Shirai, J. Mater. Chem., 2011, 21, 10407 RSC.
- M. Adachi, H. Okamura and M. Shirai, Chem. Lett., 2013, 42, 1056 CrossRef CAS.
- E. M. White, J. E. Seppala, P. M. Rushworth, B. W. Ritchie, S. Sharma and J. Locklin, Macromolecules, 2013, 46, 8882 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59 CrossRef CAS PubMed.
- D. Klinger and K. Landfester, Soft Matter, 2011, 7, 1426 RSC.
- D. Klinger and K. Landfester, Macromolecules, 2011, 44, 9758 CrossRef CAS.
- F. Ercole, H. Thissen, K. Tsang, R. A. Evans and J. S. Forsythe, Macromolecules, 2012, 45, 8387 CrossRef CAS.
- C. S. Ki, H. Shih and C.-C. Lin, Polymer, 2013, 54, 2115 CrossRef CAS PubMed.
- M. W. Tibbitt, A. M. Kloxin, L. A. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785 CrossRef CAS PubMed.
- S. Bian, J. Zheng and W. Yang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1676 CrossRef CAS.
- Q. Huang, C. Bao, W. Ji, Q. Wang and L. Zhu, J. Mater. Chem., 2012, 22, 18275 RSC.
- M. A. Azagarsamy, D. D. McKinnon, D. L. Alge and K. S. Anseth, ACS Macro Lett., 2014, 3, 515 CrossRef CAS.
- K. Suyama and H. Tachi, J. Photopolym. Sci. Technol., 2014, 27, 231 CrossRef CAS.
- K. Suyama, T. Inoue and M. Shirai, J. Photopolym. Sci. Technol., 2010, 23, 439 CrossRef CAS.
- K.-H. Song, M. Tsunooka and M. Tanaka, Makromol. Chem., Rapid Commun., 1988, 9, 519 CrossRef CAS.
- K. Suyama and M. Tsunooka, Polym. Degrad. Stab., 1994, 45, 409 CrossRef CAS.
- K. Suyama and M. Shirai, Prog. Polym. Sci., 2009, 34, 194 CrossRef CAS PubMed.
- K. Suyama, S. Ozaki and M. Shirai, React. Funct. Polym., 2013, 73, 518 CrossRef CAS PubMed.
- M. Leeman, G. Brasile, E. Gelens, T. Vries, B. Kaptein and R. Kellogg, Angew. Chem., Int. Ed., 2008, 47, 1287 CrossRef CAS PubMed.
- J. F. Cameron and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 113, 4303 CrossRef CAS.
- M. Tsunooka, S. Imono, K. Nakayama, H. Kuwabara and M. Tanaka, J. Polym. Sci., Polym. Chem. Ed., 1986, 24, 317 CrossRef CAS.
- We have confirmed the copolymerization of DBzM and TBzM with butyl acrylate or poly(ethylene glycol) monomethacrylate.
- H. B. Lueck, T. C. Swinney, B. S. Hudson and D. M. Freidrich, Chem. Phys. Lett., 1996, 258, 80 CrossRef CAS.
- R. M. Silverstein, G. Clayton and T. C. Morrill, in Spectrometric Identification of Organic Compounds, John Wiley & Sons, New York, 4th edn, 1981, p. 321 Search PubMed.
- G. Socrates, in Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons, New York, 3rd edn, 2001, p.78 Search PubMed.
- In our experimental condition, the lower conversion of the photopolymerization is suggested. In fact, for one sample film of TBzM–MA, the weight of formulation on the substrate changed from 19 mg to 10 mg on irradiation followed by removing the PET film. The weight of the film was still 10 mg after soft baking at 80 °C for 2 min, indicating that the conversion for TBzM–MA was ca. 50%.
- J. Lalevée, X. Allonas, J. P. Fouassier, H. Tachi, A. Izumitani, M. Shirai and M. Tsunooka, J. Photochem. Photobiol., A, 2002, 151, 27 CrossRef.
- T. Ohba, K. Suyama and M. Shirai, React. Funct. Polym., 2006, 66, 1189 CrossRef CAS PubMed.
- J. S. Young, A. R. Kannurpatti and C. N. Bowman, Macromol. Chem. Phys., 1998, 199, 1043 CrossRef CAS.
- J. E. Elliott and C. N. Bowman, Macromolecules, 2001, 34, 4642 CrossRef CAS.
- R. B. Fox, L. G. Isaacs, S. Stokes and R. E. Kagarise, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 2085 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00574d |
| This journal is © The Royal Society of Chemistry 2015 |