
Stabilization of silicon nanoparticles in graphene aerogel framework for lithium ion storage†
Binghui Xua,
Hao Wua,
C. X. (Cynthia) Lina,
Bo Wangab,
Zhi Zhangc and
X. S. Zhao*a
aSchool of Chemical Engineering, The University of Queensland, St. Lucia, Brisbane, QLD 4072, Australia. E-mail: george.zhao@uq.edu.au
bSchool of Chemical Engineering and Technology, Harbin Institute of Technology, Xidazhi Street, 150001 Harbin, China
cMaterials Engineering, The University of Queensland, St. Lucia, Brisbane, QLD 4072, Australia
First published on 24th March 2015
Abstract
The severe volume change and aggregation of silicon nanoparticles (SiNPs) when used as an anode for lithium ion batteries (LIBs) are the key issue. Here, we demonstrate a novel approach to wrapping SiNPs in three-dimensional reduced graphene oxide (RGO) aerogel. The RGO aerogel not only provides a porous network for entrapping SiNPs to accommodate the volume change during cycling, but also facilitates electrolyte transport. Furthermore, the continuous RGO network is favourable for electron transfer. The graphene-wrapped SiNPs were stable and displayed an excellent rate capacity, delivering a reversible capacity of about 2000 mA h g−1 after 40 cycles.
Introduction
With the fast growing demands for energy storage devices, the development of new-generation LIBs is becoming increasingly important. Achieving high capacity, excellent cycling performance and rate capability with innovative electrode materials has been one of the main challenges.1 Silicon (Si) is a promising anode material for new-generation LIBs because of its high theoretical Li ion storage capacity (∼4200 mA h g−1), significantly higher than that of commercialized graphite (372 mA h g−1). Si also features low discharge potential (∼0.4 V vs. Li/Li+), rich natural abundance, and environmental benignity.2–4 However, the realization of Si as a LIB anode has been hindered because Si suffers from not only a low intrinsic electronic conductivity but also a large specific volume changes (>300%) during lithiation and delithiation, resulting in pulverization of Si particles and electrical disconnection from the current collector, leading to a rapid capacity fading.5–7 To solve these problems, nanostructured Si electrodes such as nanoparticles,4,8–10 nanowires,11–15 nanotubes and hollow spheres16,17 have been explored. Combining SiNPs with carbon materials has also been studied.8,10,18–23Graphene, a monolayer of carbon atoms arranged in a two-dimensional (2D) honeycomb network, has been used to improve the stability and electric conductivity of nanostructured Si electrodes for LIBs.24 Due to its high electronic conductivity, superior mechanical strength and flexibility, graphene can improve electron transport and Li+ diffusion, thus enhancing the electrochemical performance of SiNPs for lithium ion storage.23,25–27 In spite of the observed improvement of the electrochemical performance of SiNPs by graphene, SiNPs still tend to aggregate. As a result, the performance of the graphene–SiNPs composites inevitably degrades during charge/discharge. Recently, three-dimensional (3D) graphene materials, including hydrogels and aerogels, have been shown to possess advantages, such as high surface area and good electrical conductivity.28–30 These 3D graphene materials may be favourable for stabilizing SiNPs.
This paper describes a method for preparing graphene-stabilized SiNPs on the basis of electrostatic interactions. Because both graphene oxide (GO) and SiNPs (there is a thin layer of SiO2 on the surface of the SiNPs) are negatively charged in a wide pH range, the SiNPs used in this work were firstly modified using poly(diallydimethylammonium chloride) (PDDA, a positively charged polyelectrolyte) to change the surface charge nature from being negative to being positive following a protocol described elsewhere.23 As schematically illustrated in Scheme 1, the PDDA-modified SiNPs interacted strongly with negatively charged GO sheets to form a Si–GO composite (hereinafter designated as Si@GO). Because of the flexibility of GO sheets, SiNPs were wrapped in by the GO sheets. The GO in the Si@GO suspension was then reduced using gallic acid (GA, a natural plant phenolic acid; see Fig. S1A† for its structure) in an oil bath at 95 °C for 4 h. It has been reported that in the presence of natural phenolic acid, GO can be reduced to assemble into RGO hydrogels driven by the enhancing hydrophobicity and π–π interactions among the nanosheets during the reduction.28,31 During this GA reduction process, yellow-brown-coloured Si@GO suspension gradually turned transparent to form a dark grey gel separating from the suspension, indicating that the GO had been reduced to reduced graphene oxide (RGO) and the SiNPs had been wrapped by the RGO architecture. In order to understand the stabilization effect of RGO sheets on SiNPs, the obtained Si@RGO hydrogel was crushed and redispersed in an aqueous GO suspension, which was further reduced with GA at 95 °C for 12 h. After freeze-drying and thermal reduction at 700 °C for 2 h under H2/Ar atmosphere, a black 3D RGO aerogel with confined SiNPs, designated as Si/RGO-AG, was obtained. For comparison purpose, another two samples were prepared. One of them, designated as Si/RGO, was prepared by mixing SiNPs with aqueous GO suspension, followed by filtration and thermal reduction at 700 °C for 2 h under H2/Ar atmosphere. The other one, designated as Si/RGO-SWAG (SiNPs were wrapped by RGO aerogel in a single step), was prepared according to the same procedure of preparing sample Si/RGO-AG except for without Step 3 (see Scheme 1).
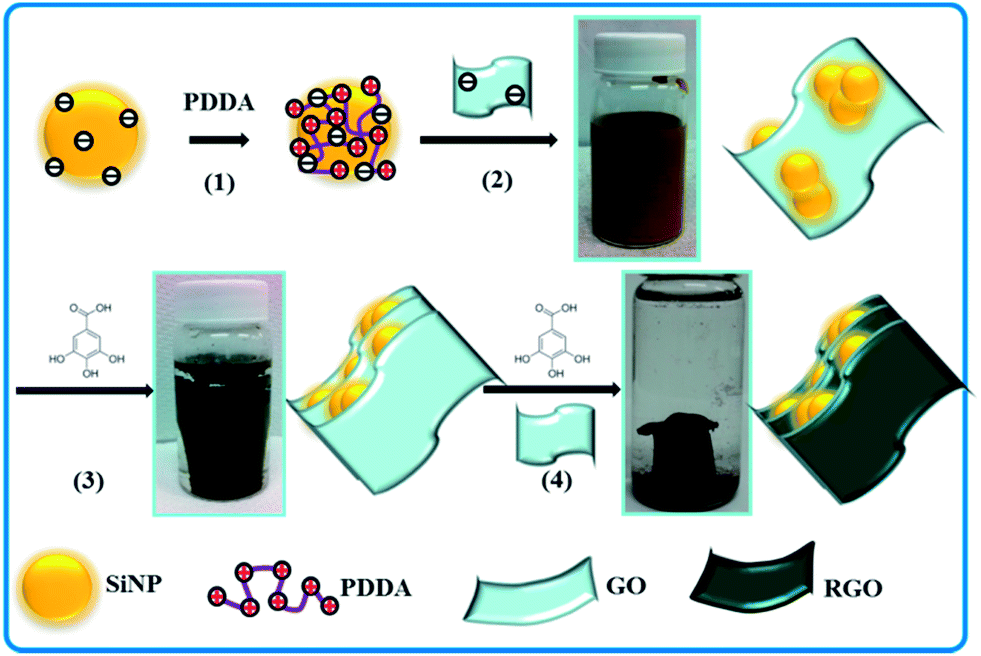 | ||
| Scheme 1 Schematic illustration of the preparation of Si/RGO-AG: (1) surface modification of SiNPs with PDDA; (2) formation of Si@GO composite via electrostatic interactions; (3) reduction of Si@GO using GA to form Si@RGO hydrogel; (4) dispersing Si@RGO in a GO suspension for further reduction using GA. | ||
Experimental section
Preparations of graphene oxide (GO)
The GO used in this work was prepared from natural graphite flake (Sigma Aldrich, 325 mesh) by using a modified Hummers method.32Preparation of RGO aerogel wrapped SiNPs (Si/RGO-AG)
SiNPs (120 mg, 99% purity, from Nanostructured & Amorphous Materials, Inc.) and 20 wt% PDDA (3.0 g, Sigma Aldrich, MW = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000) aqueous solution were dispersed in water (120 mL) under sonication in a water bath (KQ3200DE, 40 kHz). Excess PDDA was washed away by centrifugation (10000 rpm) four times, followed by vacuum drying. An aqueous GO suspension (15 mL, 2 mg mL−1) was centrifuged at 5000 rpm for 30 min to remove any graphite particles and then mixed with the above PDDA-modified SiNPs (120 mg) under sonication for 1 h. After the addition of GA (13 mg), the mixed suspension was transferred to a 20 mL capped vial and placed into an oil bath at 95 °C for 4 h to form a Si@GO hydrogel. The hydrogel was collected and redispersed in another GO aqueous suspension (15 mL, 2 mg mL−1) and GA (13 mg) under sonication. The suspension was then transferred into a 20 mL vial to prepare hydrogel at 95 °C for 12 h. The resulting hydrogel monolith was soaked in distilled water, followed by freeze-drying. Reduction was conducted in a quartz tube at 700 °C for 2 h under a H2/Ar (5:95 v/v) atmosphere with a heating rate of 2 °C min−1 to obtain sample Si/RGO-AG.
000–20000) aqueous solution were dispersed in water (120 mL) under sonication in a water bath (KQ3200DE, 40 kHz). Excess PDDA was washed away by centrifugation (10000 rpm) four times, followed by vacuum drying. An aqueous GO suspension (15 mL, 2 mg mL−1) was centrifuged at 5000 rpm for 30 min to remove any graphite particles and then mixed with the above PDDA-modified SiNPs (120 mg) under sonication for 1 h. After the addition of GA (13 mg), the mixed suspension was transferred to a 20 mL capped vial and placed into an oil bath at 95 °C for 4 h to form a Si@GO hydrogel. The hydrogel was collected and redispersed in another GO aqueous suspension (15 mL, 2 mg mL−1) and GA (13 mg) under sonication. The suspension was then transferred into a 20 mL vial to prepare hydrogel at 95 °C for 12 h. The resulting hydrogel monolith was soaked in distilled water, followed by freeze-drying. Reduction was conducted in a quartz tube at 700 °C for 2 h under a H2/Ar (5:95 v/v) atmosphere with a heating rate of 2 °C min−1 to obtain sample Si/RGO-AG.
Preparation of other samples
For comparison purpose, another two samples were prepared. One of them, designated as Si/RGO, was prepared as follows. An aqueous GO suspension (30 mL, 2 mg mL−1) was mixed with PDDA-modified SiNPs (120 mg) under sonication for 1 h. After the addition of GA (26 mg), the suspension was reduced at 95 °C in an oil bath for 12 h under magnetic stirring. Then the solid was collected by filtration, followed by vacuum drying and reduction in a quartz tube at 700 °C for 2 h under a H2/Ar (5:95 v/v) atmosphere with a heating rate of 2 °C min−1. The other sample, designated as Si/RGO-SWAG, was prepared as follows. An aqueous GO suspension (15 mL, 2 mg mL−1) was mixed with PDDA-modified SiNPs (60 mg) under sonication for 1 h. After the addition of GA (13 mg), the suspension was transferred into a 20 mL capped vial and placed in an oil bath at 95 °C for 12 h. The resulting hydrogel monolith was soaked in distilled water, followed by freeze-drying. Reduction was conducted in a quartz tube at 700 °C for 2 h under an H2/Ar (5:95 v/v) atmosphere with a heating rate of 2 °C min−1.
Characterization
The TEM and HRTEM measurement was performed on a Philips Tecnai F20 and Philips Tecnai F30 field emission transmission electron microscopes operated at 200 kV. SEM images were obtained from a JEOL 7800F scanning electron microscope operated at 5.0 kV. XRD patterns were collected on a German Bruker D8 Advanced X-ray diffractometer with Ni filtered Cu Kα radiation (40 kV, 30 mA). Thermo gravimetric analysis (TGA) was carried out on a TGA/DSC1 STARe System under air flow (25–900 °C, 5 °C min−1). X-ray photoelectron spectra (XPS) were collected on a Kratos Axis ULTRA X-ray photoelectron spectrometer using a monochromatic Al Kα (1486.6 eV) X-ray source and a 165 mm hemispherical electron energy analyzer. Nitrogen physisorption isotherms were measured at 77 K on Tristar II 3020. All samples were degassed at 200 °C for 12 h prior to the measurements. The specific surface areas of the samples were calculated using the Brunauer–Emmett–Teller (BET) method and the total pore volumes were estimated from the nitrogen volumes adsorbed at the relative pressure of 0.99. The pore size distribution curves were derived from the Barrett–Joyner–Halenda (BJH) method of isotherm analysis. Raman spectra were collected on a Thermo-Fischer Almega dispersive Raman instrument. The instrument was fitted with both 633 and 785 nm lasers. Energy-dispersive X-ray spectroscopy analysis (EDX) was conducted on a JSM6610 EDS system.Electrochemical measurements
The electrochemical properties of the prepared samples were evaluated with CR2032 coin cells. The electrodes were prepared by mixing the active materials with conductive carbon black (Super-P) and poly(vinylidene fluoride) (PVDF) dissolved in N-methyl-2-pyrrolidone (NMP) with a mass ratio of 80:10:10 to form a homogeneous slurry under magnetic stirring. The slurry was then spread onto a pure Cu foil. Pure lithium metal discs were selected as the counter electrode. The electrolyte used was 1 M LiPF6 in ethylene carbonate/dimethyl carbonate (1:1 v/v). A Celgard 2400 microporous polypropylene membrane was used as the separator. The CR2032 cells were assembled in an argon-filled glove box (Mbraun Unilab) with water and oxygen contents less than 1 ppm. Cyclic voltammetry was carried out on a CHI 660D electrochemical workstation at a scan rate of 0.1 mV s−1. The discharge and charge measurements of the batteries were performed on a Neware system in the fixed voltage window between 0.02 and 1.2 V at room temperature.
Results and discussion
The morphology and microstructure of the Si/RGO-AG composite were characterized using scanning electron microscopy (SEM), transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) techniques. The HRTEM image in Fig. S1B† shows that there was a shell around the SiNPs with a thickness of about 5 nm. The X-ray photoelectron spectroscopy (XPS) results of the SiNPs in Fig. S2† shows a peak at 103 eV, which is attributed to Si 2p electrons in Si–O4,33 confirming the shell was silicon oxide (SiO2). This SiO2 layer plays a vital role in modifying the surface chemistry of SiNPs.23 The silanol groups on the surface of the SiO2 shell can be ionized under the experimental conditions to carry a negative charge, thus enabling strong electrostatic interactions between the SiNPs and PDDA to change the SiNPs from a negatively charged surface to a positively charged surface. On the other hand, according to a recent study,34 the SiO2 layer can suppress the volume expansion of SiNPs although it consumes some lithium ions during the first discharge cycle.Fig. 1A and B are the SEM images of Si/RGO-AG under different magnifications. SiNPs with an average diameter of 100 nm dispersed the 3D RGO aerogel framework can be clearly seen. On contrast, severe aggregation of SiNPs can be observed from pure SiNPs (Fig. S3A†), sample Si/RGO (Fig. S3B†) and sample Si/RGO-SWAG (Fig. S3C and S3D†). In particular, the SiNPs in sample Si/RGO formed micro-sized aggregates. After a single-step wrapping by RGO, the aggregation of SiNPs was not significant. However, some SiNPs were not fully wrapped by RGO and aggregates can be still observed. This explains the capacity fading of sample Si/RGO-SWAG. The elemental analysis results of Si/RGO-AG (Fig. S4†) confirmed the existence of major elements Si and C and their homogeneous distribution in the 3D aerogel. In this Si/RGO-AG composite, the graphene aerogel provides a porous network for the entrapped SiNPs, thus is beneficial for accommodating the volume change of these SiNPs during electrochemical reactions. The porous network also facilitates electrolyte transport, potentially enhancing the rate capacity of LIBs. Besides, the continuous interconnected graphene network creates favourable electron pathways against cycling processes. The HRTEM image shown in Fig. 1C also clearly demonstrates the existence of a continuous RGO network with SiNPs entrapped. The HRTEM image in Fig. 1D shows that SiNPs were well-encapsulated within the RGO aerogel network.
 | ||
| Fig. 1 SEM (A and B), TEM (C), and HRTEM images (D) of Si/RGO-AG composite. | ||
Fig. 2A shows the X-ray diffraction (XRD) patterns of Si/RGO-AG and SiNPs. All diffraction peaks due to SiNPs can be seen from sample Si/RGO-AG, indicating that the silicon crystalline structure in the Si/RGO-AG composite retained after the freeze-drying and thermal reduction treatments. Fig. 2B presents the Raman spectra of Si/RGO-AG and pristine SiNPs. The absorption bands due to SiNPs can be seen from Si/RGO-AG, confirming the presence of crystalline Si particles in the composite. There are two more absorption bands at 1350 and 1596 cm−1, respectively. These two peaks are assigned to the D band and G band of graphene, respectively,35 confirming the presence of RGO in the composite.
 | ||
| Fig. 2 XRD patterns of SiNPs and Si/RGO-AG (A); Raman spectra of SiNPs and Si/RGO-AG (B); N2 adsorption/desorption isotherms (C) and BJH pore size distribution based on desorption isotherm of Si/RGO-AG (D). | ||
Fig. S5† presents the C1s XPS spectra of samples. As can be seen, GO showed three peaks at 284.9, 286.8, and 289.0 eV, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in aromatic rings, C–O–C in epoxy and alkoxy, and CO in carbonyl and carboxyl groups, respectively. It is clearly seen that the peak intensity due to C–O–C and CO bonds of sample Si/RGO-AG is significantly lower than that of sample GO. The intensity of the peak ascribed to CC however increased after reduction. These data suggest that the oxygen-containing groups on GO were largely removed, and most of the conjugated bonds were restored by GA reduction and thermal treatment.
C in aromatic rings, C–O–C in epoxy and alkoxy, and CO in carbonyl and carboxyl groups, respectively. It is clearly seen that the peak intensity due to C–O–C and CO bonds of sample Si/RGO-AG is significantly lower than that of sample GO. The intensity of the peak ascribed to CC however increased after reduction. These data suggest that the oxygen-containing groups on GO were largely removed, and most of the conjugated bonds were restored by GA reduction and thermal treatment.
The N2 adsorption/desorption isotherms of the Si/RGO-AG composite (see Fig. 2C) showed a type IV isotherm with an H3 hysteresis loop, indicating a mesoporous structure of the material. The Brunauer–Emmett–Teller (BET) surface area and pore volume of the composite was measured to be 125 m2 g−1 and 0.37 cm3 g−1, respectively, much higher than that of pure SiNPs (25 m2 g−1 and 0.055 cm3 g−1, respectively, see Fig. S6A†). The Barrett–Joyner–Halenda (BJH) pore size distribution curve of Si/RGO-AG (see Fig. 2D) demonstrates that Si/RGO-AG composite had a mesoporous structure with two pore systems. The intensive peak around 5.0 nm is due to the presence of small gaps among randomly stacked graphene sheets, while the broad peak centred at about 26 nm may be due to the large voids between inter-twisted graphene sheets.36 The high surface area along with the existence of mesopores in Si/RGO-AG should offer a large material–electrolyte contact area and promote the diffusion of Li+ ions if Si/RGO-AG is used as electrode materials for lithium storage.
Thermal gravimetric analysis (TGA) was carried out in air and the results are shown in Fig. S6B.† It can be seen that the mass of the pristine SiNPs increased slightly with a mass gain of about 0.150 wt% at 700 °C. This gain was due to the oxidation of Si by O2 to form SiO2. At 700 °C, the residual of the RGO aerogel was about 3.02 wt%, which was due to the impurities in the GO sample. The weight loss of the Si/RGO-AG composite at 700 °C was about 19.3 wt%. Based on the TGA data, the Si/RGO-AG composite was calculated to contain about 80.0 wt% SiNPs and 20.0 wt% RGO aerogel.
The electrochemical performance of the Si/RGO-AG composite as an anode was assembled and tested in a CR2032 coin cell where lithium foil was used as a counter electrode. For comparison, the cycling performance of electrode Si/RGO was also tested under the same experimental conditions. Fig. 3A shows typical cyclic voltammetry (CV) curves of electrode Si/RGO-AG in the potential range of 0.02–1.20 V (vs. Li+/Li) at a scan rate of 0.1 mV s−1 of the first two cycles, starting at the open circuit potential of 1.59 V. A broad cathodic peak in the first cycle appeared at 0.69 V, indicating the formation of solid electrolyte interphase (SEI).8 This cathodic peak disappeared in the second cycle and correlated to an initial capacity loss. The main cathodic part of the second cycle displayed a peak at 0.19 V, corresponding to the formation of Li–Si alloy phases.37 The anodic part showed two peaks at 0.34 and 0.52 V, corresponding to the phase transition from Li–Si alloys to amorphous Si.8,23
 | ||
| Fig. 3 (A) Cyclic voltammetry curves for Si/RGO-AG for the first two cycles; (B) galvanostatic discharge/charge profiles of the first three cycles; (C) cycling performance of electrode Si/RGO-AG, Si/RGO-SWAG, and Si/RGO; (D) rate capability of electrode Si/RGO-AG. | ||
Fig. 3B displays the discharge/charge profiles of the initial three cycles of electrode Si/RGO-AG under a current density of 150 mA g−1 in the voltage window of 0.02 to 1.2 V (vs. Li+/Li). The onset slope at about 0.7 V in the initial discharging curve, which disappeared in the following cycles, corresponds to the SEI formation.8 Besides, the main discharge plateau is around 0.2 V and the charge plateau is around 0.5 V. All these features are in good agreement with the CV results discussed above. The specific capacity was calculated based on the total mass of Si/RGO-AG. The initial discharge/charge capacities were 3446 and 2535 mA h g−1, respectively, to give an initial coulombic efficiency of 73.6%. The initial irreversible capacity of electrode Si/RGO-AG can be attributed to the formation SEI, the unexpected reaction of the remaining oxygen-containing groups in the RGO aerogel and SiO2 layer on the surface of SiNPs with Li ions.27,38 After the second cycle, the coulombic efficiency tended to increase and stabilize. It is interesting to note that the curves of the second and third cycles almost overlapped each other, which indicates a good cycling stability of the electrode. The reversible capacities of the second and third cycles compared with the first cycle were slightly increased, which can be attributed to the activation of the SiNPs in the Si/RGO-AG composite.
Fig. 3C shows the cycling performance of Si/RGO-AG at a current density of 150 mA g−1, together with electrodes Si/RGO-SWAG and Si/RGO. It can be seen that the initial charge and discharge capacities of electrode Si/RGO were the lowest among the three electrodes studied. This poor performance of electrode Si/RGO was probably due to severe aggregation of the SiNPs, indicating the RGO sheets did not stabilize the SiNPs well. After about 20 cycles, the reversible capacity dropped drastically to about 450 mA h g−1, confirming that the SiNPs were not stabilized well by the RGO sheets. These results suggest that the physical mixing method is not a good approach to stabilizing SiNPs. While the initial discharge/charge capacities of electrode Si/RGO-SWAG reached to 3360 and 2450 mA h g−1, respectively, its reversible capacity decreased to about 615 mA h g−1 after 40 cycles. The improved performance of electrode Si/RGO-SWAG indicates the single-step wrapping method illustrated in Scheme 1 is advantageous over the physical mixing method. This fast capacity fading however indicates that the single-step wrapping method still could not afford efficient stabilization of SiNPs. On contrast, the Si/RGO-AG electrode showed a significantly improved cycling performance with the highest initial charge/discharge capacities and delivered a reversible capacity of 1984 mA h g−1 after 40 cycles.
Fig. 3D demonstrates the rate capability of the Si/RGO-AG at current densities ranging from 170 mA g−1 to 2500 mA g−1. The battery delivered a reversible capacity of about 1000 and 600 mA h g−1 at the current densities of 2000 and 2500 mA g−1, respectively. Furthermore, the capacity reached around 2000 mA h g−1 when the current density was decreased to 170 mA g−1 after having been cycled at higher current densities, indicating a good cycling stability of Si/RGO-AG.
Fig. S7A† shows the SEM image of electrode Si/RGO-AG after 40 cycles. As can be seen, the Si/RGO-AG composite maintained its integrity and porous structure. In addition, the SiNPs were entrapped by the RGO framework, contributing to the significantly improved cycling performance of Si/RGO-AG. The Nyquist plots of the Si/RGO-AG electrode are presented in Fig. S7B.† The depressed semicircle in the high-frequency region represents the resistance of the SEI film and the charge-transfer resistance, while the straight lines in the low-frequency region corresponds to the diffusion kinetics of lithium ions. No obvious impedance increase is observed after cycling due to the stable SEI. The impedance decrease may be attributed to the gradual electrolyte transport into the electrode and the increasing conductivity of the SiNPs after lithiation.
The improved cycle performance and enhanced rate capability of Si/RGO-AG can be attributed to the following reasons: (i) the Si/RGO-AG composite created sufficient space and efficiently accommodated the drastic volume change of the entrapped SiNPs during cycling; (ii) the interconnected 3D RGO aerogel network maintained the integrity of the electrode structure, prohibited the detachment of the SiNPs from the current collector and improved the electrical conductivity of the electrode; (iii) the existence of meso- and macro- pores provided an efficient pathway for electrolyte transport and facilitated Li+ diffusion, thus enhancing the rate performance.
Conclusions
In conclusion, we have demonstrated an approach to preparing a high-performance LIB negative electrode material by encapsulating SiNPs in 3D RGO aerogel. The RGO aerogel with a porous network offers space for accommodating SiNPs, as well as facilitating electron and electrolyte transport. The composite electrode delivered a stable cycling performance with a capacity of about 2000 mA h g−1 after 40 cycles, together with an excellent rate capability.Acknowledgements
The authors would like to thank the Australia Research Council (ARC) for funding this work under projects FT100100879 and DP130101870. The facilities and technical assistance of the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis at The University of Queensland are acknowledged. The author also specially thanks the China Scholarship Council.References
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS PubMed.
- U. Kasavajjula, C. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003–1039 CrossRef CAS PubMed.
- C. M. Park, J. H. Kim, H. Kim and H. J. Sohn, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS PubMed.
- X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B. W. Sheldon and J. Wu, Adv. Energy Mater., 2014, 4, 1–23 Search PubMed.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 CAS.
- B. Scrosati, J. Hassoun and Y. K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS.
- X. S. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Chem. Commun., 2012, 48, 2198–2200 RSC.
- J. K. Lee, K. B. Smith, C. M. Hayner and H. H. Kung, Chem. Commun., 2010, 46, 2025–2027 RSC.
- S. N. Yang, G. R. Li, Q. Zhu and Q. M. Pan, J. Mater. Chem., 2012, 22, 3420–3425 RSC.
- L. F. Cui, R. Ruffo, C. K. Chan, H. Peng and Y. Cui, Nano Lett., 2008, 9, 491–495 CrossRef PubMed.
- L. F. Cui, Y. Yang, C. M. Hsu and Y. Cui, Nano Lett., 2009, 9, 3370–3374 CrossRef CAS PubMed.
- M. T. McDowell and Y. Cui, Adv. Energy Mater., 2011, 1, 894–900 CrossRef CAS.
- H. T. Nguyen, F. Yao, M. R. Zamfir, C. Biswas, K. P. So, Y. H. Lee, S. M. Kim, S. N. Cha, J. M. Kim and D. Pribat, Adv. Energy Mater., 2011, 1, 1154–1161 CrossRef CAS.
- W. L. Xu, S. S. S. Vegunta and J. C. Flake, J. Power Sources, 2011, 196, 8583–8589 CrossRef CAS PubMed.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. A. Liu, L. B. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed.
- M. H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed.
- L. W. Su, Z. Zhou and M. M. Ren, Chem. Commun., 2010, 46, 2590–2592 RSC.
- Y. Chen, C. Li, Y. G. Wang, Q. Zhang, C. Y. Xu, B. Q. Wei and L. N. An, J. Mater. Chem., 2011, 21, 18186–18190 RSC.
- M. K. Datta, J. Maranchi, S. J. Chung, R. Epur, K. Kadakia, P. Jampani and P. N. Kumta, Electrochim. Acta, 2011, 56, 4717–4723 CrossRef CAS PubMed.
- Y. H. Xu, G. P. Yin, X. Q. Cheng and P. J. Zuo, Electrochim. Acta, 2011, 56, 4403–4407 CrossRef CAS PubMed.
- Y. X. Yin, S. Xin, L. J. Wan, C. J. Li and Y. G. Guo, J. Phys. Chem. C, 2011, 115, 14148–14154 CAS.
- X. S. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Adv. Energy Mater., 2012, 2, 1086–1090 CrossRef CAS.
- C. Xu, B. Xu, Y. Gu, Z. Xiong, J. Sun and X. Zhao, Energy Environ. Sci., 2013, 6, 1388–1414 CAS.
- B. Wang, X. Li, X. Zhang, B. Luo, M. Jin, M. Liang, S. A. Dayeh, S. Picraux and L. Zhi, ACS Nano, 2013, 7, 1437–1445 CrossRef CAS PubMed.
- X. Zhao, C. M. Hayner, M. C. Kung and H. H. Kung, Adv. Energy Mater., 2011, 1, 1079–1084 CrossRef CAS.
- H. F. Xiang, K. Zhang, G. Ji, J. Y. Lee, C. J. Zou, X. D. Chen and J. S. Wu, Carbon, 2011, 49, 1787–1796 CrossRef CAS PubMed.
- W. Chen and L. Yan, Nanoscale, 2011, 3, 3132–3137 RSC.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- S.-H. Park, H.-K. Kim, D.-J. Ahn, S.-I. Lee, K. C. Roh and K.-B. Kim, Electrochem. Commun., 2013, 34, 117 CrossRef CAS PubMed.
- Z. G. Xiong, L. L. Zhang, J. Z. Ma and X. S. Zhao, Chem. Commun., 2010, 46, 6099–6101 RSC.
- V. Kesler, S. Yanovskaya, G. Kachurin, A. Leier and L. Logvinsky, Surf. Interface Anal., 2002, 33, 914–917 CrossRef CAS.
- S. Sim, P. Oh, S. Park and J. Cho, Adv. Mater., 2013, 25, 4498–4503 CrossRef CAS PubMed.
- J. Z. Wang, C. Zhong, S. L. Chou and H. K. Liu, Electrochem. Commun., 2010, 12, 1467–1470 CrossRef CAS PubMed.
- X. Zhang, Z. Sui, B. Xu, S. Yue, Y. Luo, W. Zhan and B. Liu, J. Mater. Chem., 2011, 21, 6494–6497 RSC.
- Y. Zhu, W. Liu, X. Zhang, J. He, J. Chen, Y. Wang and T. Cao, Langmuir, 2013, 29, 744–749 CrossRef CAS PubMed.
- J. Deng, H. Ji, C. Yan, J. Zhang, W. Si, S. Baunack, S. Oswald, Y. Mei and O. G. Schmidt, Angew. Chem., 2013, 125, 2382–2386 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00566c |
| This journal is © The Royal Society of Chemistry 2015 |