DOI:
10.1039/C5RA00518C
(Paper)
RSC Adv., 2015,
5, 24625-24634
Hydrothermal preparation of nest-like CuO nanostructures for non-enzymatic amperometric detection of hydrogen peroxide
Received
10th January 2015
, Accepted 2nd March 2015
First published on 3rd March 2015
Abstract
In this study, nest-like CuO nanostructures were hydrothermally synthesized at 160 °C for 6 h with 0.24 g Cu(NO3)2·2.5H2O in 20 mL H2O and 12 mL NH3·H2O without using any surfactant. The influences of the initial amount of Cu(NO3)2·2.5H2O and hydrothermal reaction time on CuO morphologies were investigated. In addition to the morphology study by scanning electron microscopy (SEM) and the crystal structure study by X-ray diffraction (XRD), as-synthesized samples were also characterized systematically by electrochemical methods including cyclic voltammetry (CV), amperometric detection (i–t) and electrochemical impedance spectroscopy (EIS). It was found that the as-prepared nest-like CuO modified glassy carbon electrode exhibited good electrochemical performance towards the reduction of H2O2. Low detection limit (0.44 μM), fast response (<2 s), and relatively high sensitivity (14.06 μA mM−1) were achieved, which was mainly due to the porous structure of the nest-like nanostructure that can provide a large specific surface area and efficient electron charge transfer and mass transport properties, thus making it a promising candidate for the efficient, stable, and precise non-enzymatic amperometric detection of H2O2.
1. Introduction
In recent years, the rapid and accurate detection of hydrogen peroxide (H2O2) is gaining more practical significance since H2O2 has been widely used in many fields including clinical treatment, food production, electronics and chemical industries, pharmaceutical and environmental analysis,1–5 etc. Compared with the various techniques for H2O2 determination,6–11 which usually involve centralized and sophisticated analytical systems, complicated processes, and expensive instruments, the electrochemical method is more attractive because of its easy fabrication and operation, fast response (real-time detection), and low cost.12,13 To date, enzyme-based electrochemical sensors have been fully developed due to their high sensitivity and extremely good selectivity. However, because of the intrinsic nature of enzymes, which are easily deformed thermally and chemically, enzyme-based sensors usually suffer from stability and reproducibility problems.14,15 Besides, they have shown certain disadvantages such as the leakage of enzymes during their immobilization and storage procedures. In addition, the preparation and purification of enzymes are usually time-consuming and expensive, and the responses of most enzyme-based sensors are not completely free from oxygen effects. Therefore, more attempts have been made to develop new enzyme-free materials for the fast, accurate, and stable detection of H2O2.
Nowadays, it has become more popular in analytical chemistry to take advantage of various metal oxide nanomaterials, because of their controllable structure, good chemical and thermal stability, high surface reaction activity and catalytic efficiency, and strong absorption ability, as well as low cost.16,17 Among them, copper oxide (CuO), a versatile p-type semiconducting material with a narrow band gap of 1.2 eV, has been extensively studied as a “future material” in potential applications such as non-enzymatic electrochemical sensors due to its excellent electrochemical and catalytic properties. It has also been widely reported that CuO nanostructures usually exhibit unique properties that are significantly different from their bulk counterpart. Therefore, a lot of attention has been paid to preparing CuO nanostructures with varied shapes, which are expected to improve their catalytic and sensing properties. For example, Weng et al. prepared CuO nanoleaf electrodes via single-step chemical oxidation of a copper foil immersed in an alkaline solution. As-synthesized CuO nanoleaf electrodes exhibited excellent non-enzymatic response to H2O2.18 Wang et al. prepared hollow copper oxide particles using the electrospinning method and modified carbon paste electrodes for H2O2 detection. High sensitivity (1746.50 μA mM−1 cm−2), low detection limits (0.022 μM) and wide linear response ranges were achieved.19 Xu et al. fabricated Cu2O–rGO nanocomposites by three different approaches, and found that the product prepared through the simple physical absorption method showed slightly better performance towards H2O2 reduction, including a wider linear range (0.03–12.8 mM), higher sensitivity (19.5 μA mM−1), and better stability.16 Zhang et al. synthesized porous Cu2O nanocubes via a sonochemical method. The sample exhibited direct electrocatalytic activity for the reduction of H2O2. Low detection limit (1.5 μM) and high sensitivity (50.6 μA mM−1) were achieved, which was mainly attributed to its porous structure.20
Based on the fact that the morphologies of the nanomaterials exert great influence on their catalytic properties, a number of physical and chemical methods have been used for the fabrication of CuO nanostructures with different morphologies, which includes electrodeposition,21 electrospinning,19 decomposition at high temperatures,22 etc. Compared with these techniques, hydrothermal processes can offer an excellent route to synthesizing various CuO nanostructures. The hydrothermal reaction is a wet chemical process with the reaction occurring at high pressures and low temperatures. It has some obvious advantages such as low temperature, simple equipment, and ease of mass production, as well as the capability to tune nano- and microstructures through simply adjusting reaction conditions.23–25 Various morphologies with large specific surface areas have been successfully synthesized using hydrothermal reactions as reported in the literature,26–30 indicating that it is a versatile and effective method. However, few papers systematically discussed the morphology evolution of CuO nanomaterials through adjusting the hydrothermal reaction conditions, as well as the detailed investigation of morphology effects on their catalytic performance towards H2O2 reduction.
In the present work, we have fabricated a non-enzymatic H2O2 electrochemical biosensor based on the nest-like CuO nanostructures via simple hydrothermal reactions without using any surfactants. The morphologies and crystal structure were characterized, and the initial amount of Cu(NO3)2 as well as hydrothermal reaction time were found to be critical to CuO morphologies. The electrochemical properties of the as-prepared sensor were studied systematically. The resulting biosensor exhibited a low detection limit, fast response, and relatively high sensitivity towards H2O2 reduction, demonstrating that the nest-like CuO modified electrode can offer great potentials for the application of electrochemical biosensor devices.
2. Experimental
2.1 Chemicals and reagents
Cu(NO3)2·2.5H2O, Na2HPO4, NaH2PO4·H2O, ascorbic acid (AA), uric acid (UA), dopamine (DA), sodium chloride (NaCl) and ammonium hydroxide were purchased from Alfa Aesar. Nafion (Nf) and potassium chloride (KCl) were obtained from Sigma-Aldrich. Hydrogen peroxide solution (H2O2, 30%) and potassium ferricyanide (K3Fe(CN)6) were purchased from Fisher Scientific. All reagents were of analytical grade and used without further purification. Phosphate buffer solution (PBS) was prepared by mixing stock solution of 0.1 M Na2HPO4 and 0.1 M NaH2PO4·H2O and adjusting the pH value with 0.1 M H3PO4 or 0.1 M NaOH. Double-distilled water was used for the preparation of buffer solution. H2O2 solution was diluted immediately before the electrochemical measurements.
2.2 Fabrication of the nest-like CuO nanostructure
In a typical procedure, 0.24 g Cu(NO3)2·2.5H2O was first dissolved into 20 mL distilled water under magnetic stirring. After 12 mL of 1 M NH3·H2O was introduced into the mixture under stirring, the clear solution was transferred into a 40 mL Teflon-lined stainless autoclave. The autoclave was sealed, maintained at 160 °C for 6 h, and then allowed to cool down to room temperature naturally. The obtained black precipitates were washed with distilled water and ethanol several times and then dried in air overnight.
2.3 Electrode modification
The nest-like CuO modified electrode was fabricated by the following way: a glassy carbon electrode (GCE, Φ = 3 mm) was first polished with a 1700# diamond paper and then washed successively with double-distilled water and ethanol several times; then, 5 μL Nf solution was diluted with 0.7 mL water and 0.3 mL isopropanol to 0.025% (v/v); next, 2 mg CuO sample was dispersed in 1 mL diluted Nf solution and followed by ultrasonication for 2 h; finally, 24 μL of the corresponding suspension (2 mg mL−1) was casted on the GCE surface and dried in air. Thus the nest-like CuO/Nf modified GCE was obtained. As-prepared modified electrode was stored in the refrigerator (4 °C) when not in use.
2.4 Apparatus and measurements
All the electrochemical experiments were carried out on a CHI 650E electrochemical analyzer (CHI, USA) with a conventional three-electrode cell. The working electrode is a bare or modified glassy carbon electrode. An Ag/AgCl and a platinum wire were used as the reference and auxiliary electrode, respectively. 0.1 M KCl was used as supporting electrolyte in 0.1 M phosphate buffer solution (pH = 7.4). A magnetic stirrer and a stirring bar were used to provide convective mass transport. The effective surface area was estimated by cyclic voltammetry in 0.1 M KCl solution containing 1 mM K3Fe(CN)6 at a variety of scan rates. Electrochemical impedance spectroscopy (EIS) measurements were performed in 0.1 M KCl solutions within the frequency range of 0.1 Hz to 10 kHz by using 5.0 mM K3Fe(CN)6–K4Fe(CN)6 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture as the electroactive probe. The electrolytic solutions were purged with purified nitrogen for at least 30 min before electrochemical measurement. All the experiments were carried out at room temperature.
:1) mixture as the electroactive probe. The electrolytic solutions were purged with purified nitrogen for at least 30 min before electrochemical measurement. All the experiments were carried out at room temperature.
Crystal phase analysis was characterized by powder X-ray diffraction (XRD) via a Bruker D2 diffractometer at the voltage of 30 kV and current of 10 mA with Cu Kα radiation (λ = 0.15418 nm), in a 2θ angular range of 20–70° at a scanning speed of 9° min−1. Surface morphologies of CuO nanostructures were observed by a scanning electron microscopy (SEM, FEI Quanta 200) operating at 5 kV and 20 kV, respectively.
3. Results and discussion
3.1 Microstructure characterization of the nest-like CuO nanostructure
The surface morphology of the nest-like CuO nanostructure was characterized by SEM and the image is shown in Fig. 1(a). The magnified SEM image was displayed in Fig. 1(b). It can be clearly observed that the entire architecture of the nest-like CuO nanostructure was constructed by randomly oriented nanoplatelets, with a lot of pores on the surface, which can provide a large specific surface area. As a result, this nanostructure ensured high surface reaction activity by providing efficient transmission channels for the analyte molecules to reach the active sites,30 thus beneficial for improving the sensitivity and stability of the modified electrode.
 |
| | Fig. 1 The SEM images (a and b) and XRD pattern (c) of the nest-like CuO nanostructure. | |
Fig. 1(c) exhibits the typical XRD pattern of the as-prepared nest-like CuO nanostructure. All the diffraction peaks can be indexed to the monoclinic phase of CuO (tenorite phase; space group C2/c; with lattice constants of a = 4.6853 Å, b = 3.4257 Å, c = 5.1303 Å, and β = 99.5490°) and no impurity peaks were detected compared with the standard diffraction peaks (ICDD-PDF card no. 00-045-0397), indicating high purity of the as-prepared CuO sample. Meanwhile, the sharp diffraction peaks indicated high crystallinity of the product that was obtained through the simple hydrothermal reaction.
3.2 Formation mechanism of the nest-like CuO nanostructure
Based on the experimental results, we have studied the determining factors of forming nest-like CuO nanostructures and proposed the formation mechanism. The overall reactions involved in the growth of CuO crystals in the experiment were considered as follows:31,32| | |
Cu2+ + 4NH3 → [Cu(NH3)4]2+
| (1) |
| | |
[Cu(NH3)4]2+ + 2OH− → Cu(OH)2 + NH4OH
| (2) |
When ammonium hydroxide was added into the Cu(NO3)2 solution under magnetic stirring, the Cu2+ ions reacted with NH3 immediately to form complex [Cu(NH3)4]2+ ions according to eqn (1). Large amounts of [Cu(NH3)4]2+ were formed as time elapsed, which were stabilized by a strong Jahn–Teller effect.33,34 These initially formed complex ions were considered as the basic building units for the formation of CuO nanostructures. During the hydrothermal process, the as-formed complex [Cu(NH3)4]2+ ions tended to react with the hydroxyl ions in the precursor solution, leading to the formation of Cu(OH)2 as shown in eqn (2). The initially formed Cu(OH)2 would directly dehydrate to CuO nuclei with the loss of one water molecule according to eqn (3). Thus a large amount of CuO nuclei were generated simultaneously and they collided with each other, leading to crystal growth. The primarily formed CuO crystals were energetically favored on their high-energy surfaces and would aggregate to form large crystals that can greatly reduce the interfacial energy. Due to the anisotropic growth of monoclinic CuO crystal35,36 and the oriented attachment mechanism,37 these CuO nuclei were able to form the platelet-like crystals along a preferred axis orientation. Finally, the formed nanoplatelets were self-assembled in a circular fashion to build CuO microspheres.
If the amount of Cu(NO3)2 in the precursor solution was small (0.048 g), then there would not be enough self-assembled microspheres for further growth. In this situation the final products obtained from hydrothermal reaction were the primarily formed microspheres with rough surfaces, as well as a small number of partially combined dumbbell-like CuO, which can be observed in Fig. 2(a). On the other hand, when too much Cu(NO3)2 was used (0.48 g) in the precursor, the number of CuO microspheres would also increase dramatically, and they would assemble compactly to form the aggregate structure, as exhibited in Fig. 2(b).
 |
| | Fig. 2 Different morphologies of CuO nanostructures by using different amounts of Cu(NO3)2 precursors: (a) 0.048 g, and (b) 0.48 g; and different hydrothermal reaction time: (c) 0.5 h, (d) 1 h, (e) 2 h, (f) 4 h, and (g) 8 h. Figure (h) shows XRD patterns of CuO samples prepared with 0.5–8 h hydrothermal reaction times. | |
In order to investigate the growth process of the nest-like CuO nanostructure, time-dependent experiments were also carried out by hydrothermal reactions at 160 °C with different reaction times (0.5–8 h). As can be observed from Fig. 2(c)–(e), at the initial stage of reaction, only CuO microspheres were formed. After increasing the reaction time from 0.5 to 2 h, the amount of CuO microspheres increased gradually, and they showed the tendency to collide with each other. Fig. 2(f) exhibits the SEM image of the product obtained after 4 h. It was clearly seen that the as-formed microspheres combined with each other. From the inset magnified SEM image we can observe the adjacent region between each two microspheres, which clearly indicated the combination of CuO microspheres. When the autoclave was maintained at 160 °C for 6 h, the nest-like CuO nanostructure was observed as shown in Fig. 1(a) and (b). The adjacent combination regions among different microspheres cannot be clearly seen, and the porous structure on the surface formed, leading to the nest-like morphology. After further increasing the reaction time to 8 h, the nest-like structure disappeared, leaving a dense and compacted surface morphology as shown in Fig. 2(g). Fig. 2(h) shows XRD patterns of the samples prepared with 0.5, 1, 2, 4 and 8 h reaction times. All the samples exhibit the same crystal structure as the nest-like CuO (monoclinic phase, ICDD-PDF card no. 00-045-0397), indicating that there is no crystal structure change after increasing the reaction time. Therefore, it is believed that the appropriate amount of Cu(NO3)2 precursor and suitable hydrothermal reaction time are necessary for the formation of nest-like CuO nanostructures.
3.3 Electrochemical impedance spectroscopy (EIS)
The electrochemical impedance spectroscopy is an effective method for probing the electron charge transfer properties between the electrolyte and the electrode surface. In this study, EIS was employed to investigate the electron charge transfer resistance (Ret) of the nest-like CuO modified glassy carbon electrode (GCE). A 10 mL 0.1 M KCl solution containing 5.0 mM K3Fe(CN)6/K4Fe(CN)6 (1:1) was used as the supporting electrolyte. The applied frequency was in the range of 0.1 Hz to 10 kHz under an open circuit potential of +0.22 V. As shown in Fig. 3(a), the bare GCE exhibited an almost straight line at the low frequency region which corresponded to the diffusion process and a small semicircle at the high frequency region which was associated with the electron charge transfer process. The Ret was estimated to be about 500 Ω from the diameter of the semicircle. After coating Nafion on the GCE surface, a dramatic increase of the interfacial Ret can be observed from the inset in Fig. 3(b) (≈24 kΩ), which was because Nafion formed an insulating layer on the electrode surface and thus obstructed the electron charge transfer process. However, when the nest-like CuO was loaded onto the Nafion membrane, the Ret decreased obviously (≈2300 Ω, Fig. 3(c)), which proved that the incorporation of the nest-like CuO facilitated the electron charge transfer process and therefore can be beneficial for the H2O2 sensing capabilities.
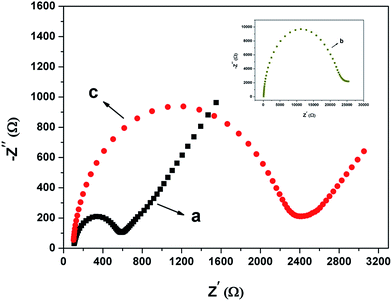 |
| | Fig. 3 Nyquist plots of EIS in 0.1 m KCl solutions containing 5.0 mm K3Fe(CN)6/K4Fe(CN)6 (1:1) for (a) bare GCE, (b) Nf/GCE, and (c) nest-like CuO modified GCE. | |
3.4 Determination of electrochemical effective surface area
The electrochemical effective surface area of the nest-like CuO modified electrode was estimated by cyclic voltammetry using ferricyanide as a redox probe. Fig. 4(a) exhibits a series of cyclic voltammograms recorded as a function of different scan rates (10–300 mV s−1) in 1 mM K3Fe(CN)6 solution with 0.1 M KCl as the supporting electrolyte. The dependence of the peak current on the square root of the scan rate is shown in Fig. 4(b). It can be clearly seen that the peak current increases linearly with the increase of the square root of the scan rate, which demonstrated that the reaction occurred on the CuO modified glassy carbon electrode was nearly reversible, and the mass transfer process in the double layer region of the electrode surface was mainly controlled by diffusion.
 |
| | Fig. 4 (a) CVs of the nest-like CuO modified GCE in a 1 mm ferricyanide solution containing 0.1 m KCl at different scan rates (10–300 mV s−1), and (b) the schematic diagram of the peak current as a function of the square root of the scan rate for determination of the electrochemical effective surface area. | |
The quantitative relationship between peak currents and scan rates can be described using Randles–Sevcik equation under semi-infinite linear diffusion conditions and room temperature (25 °C):17
| | |
Ip = (2.69 × 105)n3/2AD01/2C0ν1/2
| (4) |
where
Ip is the peak current,
n is the number of electrons participating in the redox reaction (
n = 1),
A is the electrochemical effective surface area (cm
2),
D0 is the diffusion coefficient of the molecules in solution (
D0 = 7.6 × 10
−6 cm
2 s
−1),
C0 is the concentration of the probe molecule in solution (mol cm
−3), and
ν is the scan rate (V s
−1). Therefore, the effective surface area can be calculated from the value of the slope of
Ip ∼
ν1/2, since
n,
D0 and
C0 are constant. The linear relationship between
Ip and
ν1/2 can be expressed as follows:
Ip = (3.7123 × 10
−4)
ν1/2 – 2.19026 × 10
−5 (
A, V s
−1,
R = 0.99894), and the effective surface area of the nest-like CuO modified electrode was calculated to be 0.5006 cm
2. Moreover, the effective surface area for the bare glassy carbon electrode was 0.1435 cm
2 according to the literature.
38 Therefore, the above result indicated that the electrode effective surface area increased obviously after the electrode was modified with the nest-like CuO nanostructure, which was able to enhance the current response towards H
2O
2 on the electrode surface.
3.5 Cyclic voltammetry study of the nest-like CuO modified electrode towards H2O2
The electrochemical performance of the nest-like CuO modified GCE was investigated by cyclic voltammetry (CV). Fig. 5 shows the typical cyclic voltammograms of the bare GCE, Nf/GCE, and nest-like CuO modified GCE in oxygen-free 0.1 M PBS (pH = 7.4) containing 0.1 M KCl. Clearly no obvious electrochemical redox peaks were observed when CVs were performed on bare GCE and Nf/GCE (Fig. 5(a) and (b)). After the nest-like CuO nanostructure was used to modify the electrode, a pair of well-defined redox peaks can be observed under the same experimental conditions (Fig. 5(c)), which was ascribed to the reversible reduction and oxidation of the CuII/CuI couple in CuO.
 |
| | Fig. 5 Cyclic voltammograms of (a) bare GCE, (b) Nf/GCE, and (c) nest-like CuO modified GCE in deoxygenized 0.1 m PBS (pH = 7.4) containing 0.1 m KCl. Scan rate: 100 mV s−1. | |
In order to investigate the application of the nest-like CuO modified electrode in amperometric biosensing, its electrocatalytic activity towards reduction of H2O2 was also examined by the cyclic voltammetry, which is a convenient and efficient technique for characterization of the electrocatalytic activity of materials. As shown in Fig. 6, when 1 mM H2O2 was added into the 0.1 M PBS (pH = 7.4) containing 0.1 M KCl, an obvious increase of the reduction peak current was observed in the deoxygenized environment compared with the system without H2O2. The increase of the reduction peak intensity became greater with further increasing the H2O2 concentration from 1.0 to 2.0 mM. Therefore, the above experimental results indicated that the nest-like CuO modified electrode exhibited excellent electrocatalytic activity towards H2O2. The catalytic mechanism for the reduction of H2O2 can be illustrated as follows: Cu(II) was first electrochemically reduced to Cu(I), which then reacted chemically with H2O2 and resulted in the H2O2 reverted to H2O and in the regeneration of the catalyst.39
 |
| | Fig. 6 Cyclic voltammograms of the nest-like CuO modified electrode in deoxygenized 0.1 m PBS (pH = 7.4) containing 0.1 m KCl with different concentrations of H2O2: (a) 0.0 mm, (b) 1.0 mm (c) 2.0 mm. Scan rate: 100 mV s−1. | |
3.6 Effect of the scan rate
In order to further investigate the electrochemical properties of the as-prepared sample, the effect of the scan rate on the voltammetric behavior was studied. Fig. 7(a) shows the CVs of the nest-like CuO modified electrode in 0.1 M PBS (pH = 7.4) containing 0.1 M KCl at different scan rates in the range from 20 to 200 mV s−1. It can be clearly seen that after increasing scan rates, both cathodic and anodic peak currents increased gradually, but the peak potentials shifted to opposite directions, i.e. negatively for the cathodic peak and positively for the anodic peak. Besides, both the cathodic and anodic peak currents increased linearly with the increase of the square root of the scan rate as demonstrated in Fig. 7(b), which further confirmed that the kinetics of the overall process was mainly controlled by the diffusion process. The regression equations were expressed as Ipa = −0.6589ν1/2 + 1.5871 (μA, mV s−1, R = 0.99779) and Ipc = 0.5494ν1/2 − 0.3282 (μA, mV s−1, R = 0.99903), respectively.
 |
| | Fig. 7 (a) Cyclic voltammograms of the nest-like CuO modified electrode in deoxygenized 0.1 m PBS (pH = 7.4) containing 0.1 m KCl at various scan rate (20–200 mV s−1); (b) plots of anodic and cathodic peak currents versus the square root of the scan rate: (c) dependence of anodic and cathodic peak potentials versus the logarithm of the scan rate. | |
The relationship between the peak potentials (Epa) and the logarithm of scan rate (logν) was shown in Fig. 7(c). Two straight lines were yielded with the slopes of −2.303RT/αnF and 2.303RT/(1 − α)nF for cathodic peak and anodic peak, respectively, where α is the electron transfer coefficient, n is the number of exchange electrons (n = 1), R, T, and F have their usual meaning. Two linear regression equations Epa = 0.02906 logν + 0.01722 (V, mV s−1, R = 0.9955) and Epc = −0.08674 logν + 0.03528 (V, mV s−1, R = 0.90901) can be obtained corresponding to anodic and cathodic peak potentials, respectively. So the value of α can be estimated to be 0.91 from the slopes of the straight lines based on the following equation:30,40
| |
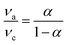 | (5) |
where
νa and
νc were determined by the
x-intercepts of the lines for the anodic and cathodic branches, respectively. The minimum value of
α should be 0.5 for all standard reaction mechanism.
41 The value at approximately 0.5 indicated that there were equal possibilities that the reaction activated transition state can form either products or reactants.
42 Thus, the value larger than 0.5 indicated a more favored reaction mechanism,
41 which explained why the electrocatalytic reduction of H
2O
2 was more favored on the nest-like CuO modified electrode compared with the bare glassy carbon electrode.
3.7 Amperometric response of H2O2 on the nest-like CuO modified electrode
Fig. 8(a) shows the steady state amperometric response of the nest-like CuO modified electrode with successive additions of H2O2 into continuously stirred 0.1 M PBS solution (pH = 7.4) containing 0.1 M KCl at an applied potential of −0.2 V. Since there was no obviously amperometric response to dissolved oxygen in the buffer solution on modified electrode, the hydrogen peroxide can be determined without inhibition of oxygen. It was clearly seen that the modified electrode exhibited a fast and sensitive response towards H2O2. The steady-state current of as-prepared sensor reached another steady-state value (95% of the maximum) in less than 2 s after the addition of H2O2. In Fig. 8(b) we can observe that the amperometric current increased with increasing the concentration of H2O2. A good linear relationship can be obtained in the range from 2.5 to 300 μM with the calculated detection limit of 0.44 μM at a signal-to-noise ratio of 3. The sensitivity of the proposed biosensor was estimated to be 14.06 μA mM−1, which is apparently larger than the CuO samples prepared with 4 and 6 h hydrothermal reaction times (the sensitivities were calculated to be 11.71 and 10.94 μA mM−1 respectively). The electrochemical performance of the as-prepared biosensor in this research was also compared with other sensors reported in the literature, and is shown in Table 1. As can be observed, the as-prepared nest-like CuO modified electrode exhibited excellent catalytic performance, i.e. a low detection limit, fast response, and relatively high sensitivity, which was due to its porous structure on the surface, large specific surface area, and efficient electron charge transfer and mass transport properties, thus making it an excellent platform for the efficient and sensitive detection of H2O2.
 |
| | Fig. 8 (a) A typical current–time curve of the nest-like CuO modified electrode upon successive additions of H2O2 into gently stirred 0.1 m PBS (pH = 7.4) containing 0.1 m KCl at an applied potential of −0.2 V, and (b) linear dependence of the amperometric response on the H2O2 concentration. | |
Table 1 Comparison of various nanomaterial-based biosensors for H2O2 detection
| Modified materials |
Sensitivity (μA mM−1) |
Linear range (μM) |
Detection limit (μM) |
Response time (s) |
References |
| Nest-like CuO |
14.06 |
2.5–300 |
0.44 |
<2 |
This work |
| Cu2O |
8.7 |
10–45 |
2.6 |
<2 |
16 |
| CuO nanoflower |
22.09 |
42.5–40000 |
— |
<10 |
17 |
| CuO nanoleaf |
11 |
96–8880 |
25 |
<5 |
18 |
| Cu2O microcubes |
50.6 |
5–1500 |
1.5 |
<3 |
20 |
| Flower like CuO |
— |
5–180 |
1.6 |
<3 |
43 |
| CuO-Tyr |
2.72 |
100–36000 |
2 |
— |
44 |
| Cu2O/graphene |
— |
300–7800 |
20.8 |
<7 |
45 |
| CuO nanoplates |
0.265 |
500–4500 |
— |
<10 |
46 |
3.8 Stability, reproducibility and selectivity
The stability of the nest-like CuO modified electrode was examined by measuring its remaining current response to 1 mM H2O2. The CV peak current of the modified electrode retained 93.7% of its initial value after stored at 4 °C in a refrigerator for one week. In the next three weeks the peak current response still retained 86.2% of its initial value. The good stability came from the Nafion membrane, which exhibited good chemical stability that can enhance the stability of CuO nanostructures. The unique nest-like structure prepared in our experiment can also contribute to the long-term stability of the modified electrode. The reproducibility was investigated by comparing the amperometric current responses to H2O2 of five modified electrodes that were prepared independently. The relative standard deviation (R.S.D.) was about 2.8% for the nest-like CuO modified electrode, indicating acceptable reproducibility for the modified electrode.
Selectivity is also very important for the practical application of biosensors. Therefore the selectivity of the nest-like CuO modified electrode was evaluated by using four interfering substances: ascorbic acid (AA), uric acid (UA), dopamine (DA), and NaCl, all at 0.1 mM levels. The amperometric response of the biosensor upon successive injections of 0.1 mM H2O2 and 0.1 mM interfering species is exhibited in Fig. 9. It was clearly shown that as-prepared sensor exhibited little response to AA and DA, and almost negligible response to UA and NaCl. This was probability due to the low working potential, as well as the use of Nafion, which was negatively charged and able to repel the interfering substances such as ascorbic acid and uric acid easily.37 Therefore, the as-prepared sensor exhibited high selectivity towards the detection of H2O2.
 |
| | Fig. 9 Current response of the nest-like CuO modified electrode at −0.2 V for 0.1 mm H2O2 in the presence of 0.1 mm ascorbic acid (AA), uric acid (UA), dopamine (DA), and NaCl. | |
4. Conclusions
In this work, nest-like CuO nanostructures were obtained through a simple hydrothermal reaction without using any surfactants. The influences of the initial amount of Cu(NO3)2 and hydrothermal reaction time on the morphology change of CuO nanostructures were discussed. A possible formation mechanism of the nest-like CuO nanostructure was proposed. Non-enzymatic amperometric biosensor was fabricated based on the nest-like CuO modified glassy carbon electrode, and its electrochemical performance towards H2O2 reduction was evaluated. The experimental results indicated that the as-prepared sensor exhibited excellent electrochemical catalytic performance towards H2O2, including a low detection limit (0.44 μM), fast response (<2 s), relatively high sensitivity (14.06 μA mM−1), good stability, and favorable selectivity. This was mainly due to its porous structure on the surface, which can provide a large specific surface area and efficient analyte transmission channels, contributing to its higher surface reaction activity and efficient electron charge transport properties. The use of Nafion also provided good chemical stability and anti-interference ability. In summary, the simple preparation, easy electrode modification, and excellent electrochemical catalytic performance made the nest-like CuO/Nf based-sensor one of the promising candidates for the electrochemical detection of hydrogen peroxide.
Notes and references
- S. J. Yao, J. H. Xu, Y. Wang, X. X. Chen, Y. X. Xu and S. S. Hu, Anal. Chim. Acta, 2006, 557, 78–84 CrossRef CAS PubMed.
- O. S. Wolfbeis, A. Dürkop, M. Wu and Z. H. Lin, Angew. Chem., Int. Ed., 2002, 41, 4495–4498 CrossRef CAS.
- P. Gao and D. W. Liu, Sens. Actuators, B, 2015, 208, 346–354 CrossRef CAS PubMed.
- H. Elzanowska, E. Abu-Irhayem, B. Skrzynecka and V. I. Birss, Electroanalysis, 2004, 16, 478–490 CrossRef CAS.
- A. A. Karyakin, E. E. Karyakina and L. Gorton, Anal. Chem., 2000, 72, 1720–1723 CrossRef CAS.
- Z. H. Li, D. H. Li, K. Oshita and S. Motomizu, Talanta, 2010, 82, 1225–1229 CrossRef CAS PubMed.
- T. Jiao, B. D. Leca-Bouvier, P. Boullanger, L. J. Blum and A. P. Girard-Egrot, Colloids Surf., A, 2008, 321, 143–146 CrossRef CAS PubMed.
- M. L. Di Paolo, M. Scarpa and A. Rigo, J. Biochem. Biophys. Methods, 1994, 28, 205–214 CrossRef CAS.
- N. V. Klassen, D. Marchington and H. C. E. Mcgowan, Anal. Chem., 1994, 66, 2921–2925 CrossRef CAS.
- K. Cui, Y. H. Song, Y. Yao, Z. Z. Huang and L. Wang, Electrochem. Commun., 2008, 10, 663–667 CrossRef CAS PubMed.
- N. N. Wei, X. Xin, J. Y. Du and J. L. Li, Biosens. Bioelectron., 2011, 26, 3602–3607 CrossRef CAS PubMed.
- C. M. Welch, C. E. Banks, A. O. Simm and R. G. Compton, Anal. Bioanal. Chem., 2005, 382, 12–21 CrossRef CAS PubMed.
- C. Batchelor-McAuley, Y. Du, G. G. Wildgoose and R. G. Compton, Sens. Actuators, B, 2008, 135, 230–235 CrossRef CAS PubMed.
- K. E. Toghill and R. G. Compton, Int. J. Electrochem. Sci., 2010, 5, 1246–1301 CAS.
- C. X. Lei, S. Q. Hu, G. L. Shen and R. Q. Yu, Talanta, 2003, 59, 981–988 CrossRef CAS.
- F. G. Xu, M. Deng, G. Y. Li, S. H. Chen and L. Wang, Electrochim. Acta, 2013, 88, 59–65 CrossRef CAS PubMed.
- M. J. Song, S. W. Hwang and D. M. Whang, Talanta, 2010, 80, 1648–1652 CrossRef CAS PubMed.
- S. H. Weng, Y. J. Zheng, C. F. Zhao, J. Z. Zhou, L. Q. Lin, Z. F. Zheng and X. H. Lin, Microchim. Acta, 2013, 180, 371–378 CrossRef CAS.
- B. J. Wang, L. Q. Luo, Y. P. Ding, D. S. Zhao and Q. L. Zhang, Colloids Surf., B, 2012, 97, 51–56 CrossRef CAS PubMed.
- L. Zhang, H. Li, Y. H. Ni, J. Li, K. M. Liao and G. C. Zhao, Electrochem. Commun., 2009, 11, 812–815 CrossRef CAS PubMed.
- X. Y. Song, S. X. Sun, W. M. Zhang, H. Y. Yu and W. L. Fan, J. Phys. Chem. B, 2004, 108, 5200–5205 CrossRef CAS.
- R. V. Kumar, R. Elgamiel, Y. Diamant and A. Gedanken, Langmuir, 2001, 17, 1406–1410 CrossRef CAS.
- Q. Liu, H. J. Liu, Y. Y. Liang, Z. Xu and G. Yin, Mater. Res. Bull., 2006, 41, 697–702 CrossRef CAS PubMed.
- D. Li, Y. H. Leung, A. B. Djurisic, Z. T. Liu, M. H. Xie, J. Gao and W. K. Chan, J. Cryst. Growth, 2005, 282, 105–111 CrossRef CAS PubMed.
- H. M. Xiao, S. Y. Fu, L. P. Zhu, Y. Q. Li and G. Yang, Eur. J. Inorg. Chem., 2007, 14, 1966–1971 CrossRef.
- S. L. Wang, H. Xu, L. Q. Qian, X. Jia, J. W. Wang, Y. Y. Liu and W. H. Tang, J. Solid State Chem., 2009, 182, 1088–1093 CrossRef CAS PubMed.
- M. A. Dara, Y. S. Kim, W. B. Kim, J. M. Sohn and H. S. Shin, Appl. Surf. Sci., 2008, 254, 7477–7481 CrossRef PubMed.
- Y. C. Zhao, X. Y. Song, Z. L. Yin and Q. S. Song, J. Colloid Interface Sci., 2013, 396, 29–38 CrossRef CAS PubMed.
- F. Teng, W. Q. Yao, Y. F. Zheng, Y. T. Ma, Y. Teng, T. G. Xu, S. H. Liang and Y. F. Zhu, Sens. Actuators, B, 2008, 134, 761–768 CrossRef CAS PubMed.
- L. Cui, H. S. Yin, J. Dong, H. Fan, T. Liu, P. Ju and S. Y. Ai, Biosens. Bioelectron., 2011, 26, 3278–3283 CrossRef CAS PubMed.
- J. Liu, J. Jin, Z. Deng, S. Z. Huang, Z. Y. Hu, L. Wang, C. Wang, L. H. Chen, Y. Li, G. V. Tendeloo and B. L. Su, J. Colloid Interface Sci., 2012, 384, 1–9 CrossRef CAS PubMed.
- X. F. Guan, L. P. Li, G. S. Li, Z. W. Fu, J. Zheng and T. J. Yan, J. Alloys Compd., 2011, 509, 3367–3374 CrossRef CAS PubMed.
- Y. Cudennec, A. Lecerf and Y. Gérault, Eur. J. Solid State Inorg. Chem., 1995, 32, 1013–1022 CAS.
- Y. Cudennec, A. Riou, A. Lecerf and Y. C. R. Gérault, Acad. Sci. Paris, Ser. Iic., 2000, 3, 661–666 CAS.
- G. Z. Shen and D. Chen, J. Am. Chem. Soc., 2006, 128, 11762–11763 CrossRef CAS PubMed.
- X. Q. Wang, G. C. Xi, S. L. Xiong, Y. K. Liu, B. J. Xi, W. C. Yu and Y. T. Qian, Cryst. Growth Des., 2007, 7, 930–934 CAS.
- M. A. Dar, Q. Ahsanulhaq, Y. S. Kim, J. M. Sohn, W. B. Kim and H. S. Shin, Appl. Surf. Sci., 2009, 255, 6279–6284 CrossRef CAS PubMed.
- L. Cui, J. Y. Zhu, X. M. Meng, H. S. Yin, X. P. Pan and S. Y. Ai, Sens. Actuators, B, 2012, 161, 641–647 CrossRef CAS PubMed.
- X. M. Miao, R. Yuan, Y. Q. Chai, Y. T. Shi and Y. Y. Yuan, J. Electroanal. Chem., 2008, 612, 157–163 CrossRef CAS PubMed.
- A. L. Eckermann, D. J. Feld, J. A. Shaw and T. J. Meade, Coord. Chem. Rev., 2010, 254, 1769–1802 CrossRef CAS PubMed.
- B. O. Agboola and T. Nyokong, Anal. Chim. Acta, 2007, 587, 116–123 CrossRef CAS PubMed.
- J. N. Soderberg, A. C. Co, A. H. C. Sirk and V. I. Birss, J. Phys. Chem. B, 2006, 110, 10401–10410 CrossRef CAS PubMed.
- L. Zhang, F. F. Yuan, X. H. Zhang and L. M. Yang, Chem. Cent. J., 2011, 5, 75–83 CrossRef CAS PubMed.
- M. U. Anu Prathap, B. Kaur and R. Srivastava, J. Colloid Interface Sci., 2012, 370, 144–154 CrossRef CAS PubMed.
- M. M. Liu, R. Liu and W. Chen, Biosens. Bioelectron., 2013, 45, 206–212 CrossRef CAS PubMed.
- R. Li, J. M. Du, Y. X. Luan, Y. G. Xue, H. Zou, G. S. Zhuang and Z. H. Li, Sens. Actuators, B, 2012, 168, 156–164 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 









