DOI:
10.1039/C5RA00450K
(Paper)
RSC Adv., 2015,
5, 17967-17975
Morphology development of PP/POE blends with high loading of magnesium hydroxide
Received
9th January 2015
, Accepted 29th January 2015
First published on 2nd February 2015
Abstract
The influence of a high loading of magnesium hydroxide (Mg(OH)2, MDH) on the morphology and properties of polypropylene (PP)/ethylene–octene copolymer (POE) blends has been investigated via scanning electron microscopy, dynamic mechanical thermal analysis and tensile mechanical testing. It was demonstrated that the mechanical properties, especially the elongation at break, are highly related to the phase structure exhibited by the composites. In the PP/POE 90/10 and 70/30 blends, the addition of a high loading of MDH lowered the average diameter of the dispersed POE domains, also the MDH and POE domains were separately dispersed in the PP matrix. Meanwhile, the elongation at break of the samples sharply declined to an unacceptable level. While in the PP/POE 50/50 blends, a co-continuous structure was formed and it could be maintained even after a large amount of MDH was added. The co-continuous structure was found to be a key factor for tolerating high loading of additives and retaining acceptable mechanical properties, especially the elongation at break.
Introduction
Polymer blending is used extensively because it is a relatively easy way to develop new materials that exhibit a favorable combination of properties from well-developed polymers, depending on the selection of blend components. To achieve the desirable properties, the key is to properly control the phase morphology and their phase sizes in the blends. The dispersion and microstructure formed during processing may be affected by several factors, such as composition, processing conditions, and the inherent properties of each component.1 Considering that most polymers are immiscible, multifarious morphologies can be formed under appropriate conditions. Among these morphologies, the droplet-in-matrix morphology is the most common type compared with fibrillar, lamellar, and other morphologies. Especially for rubber toughened polymers, a large amount of literature has been published focusing on the influence of rubber size, rubber type, and compatibilizers on the mechanical properties. Recently, co-continuous morphology is garnering much attention due to the better combination of component properties compared to the blends with a droplet-in-matrix morphology, which leads to a potential enhancement of mechanical, optical, conductive, and transport properties.2–13 Generally, if two polymers are processed under proper conditions of composition and viscosity ratio, it may be possible to cause the two components to form co-continuous interlocking phases regardless of miscibility, which is called phase co-continuity. In a typical dual phase co-continuous structure, two phases interpenetrate with each other, suggesting that both phases remain continuous throughout the material,9,11,13 and the resulting microstructure in such blends enables each phase to share in the load bearing capability of the material.2,3,13 This somewhat reduces the need for efficient stress transfer between the phases required in dispersed phase blends; and this type of morphology has been observed in various polymer blends under the proper conditions. Varied research has been done to investigate the co-continuous structured polymer composites via various techniques, including the solvent extraction technique, microscopy, and rheological analysis.11,13–29 Besides measurements like dynamic mechanical analysis (DMTA), tests such as stress–strain behavior, impact properties, and electrical conductivity or resistivity are useful in distinguishing between disperse and co-continuous structures.9–11 However, the mechanism of formation of co-continuous structures during processing, and whether a co-continuous structure is stable or an intermediate step that eventually transforms into dispersed morphology, is not yet fully understood or clearly described. Macosko et al. proposed a sheet-forming mechanism that elongated sheet or droplets of the dispersed phase should be formed during the melt mixing process.24,25 Favis et al. summarized that the co-continuous morphology should be the result of the coalescence of such elongated threads under proper conditions.28,29 Willemse et al. investigated the relation between interfacial tension and the composition range of co-continuity in the uncompatibilized blends. The critical composition for full co-continuity is found to increase with increasing interfacial tension and the co-continuous composition range is narrowed.11,13 The lower the interfacial tension, the more stable the elongated droplets.27 Adding compatibilizers is a common method to realize the reduction in interfacial tension, this method, however, would lead to other effects on phase structures. Leal et al. suggested that, compared with the breaking up of the minor phase domains, the major effect of the copolymers on the interface is to inhibit the coalescence between the dispersed particles.30–32 It is shown by several authors that much finer co-continuous structures could be achieved after adding compatibilizers.33–35 However, since high coalescence is required of the dispersed particles to form a co-continuous morphology, adding compatibilizers will further narrow down, by decreasing the probability of coalescence, the composition range to form a co-continuous structure in an immiscible system.36,37 Willis et al. observed a narrowed composition interval of the co-continuous structures when adding polyethylene-based ionomer into the polypropylene/polyamide blends.38 It is also reported by Zhang et al. that the addition of PS-graft-PA6 narrowed the co-continuous interval of the PS/PA6 blends, particularly from 40/60–65/35 (without compatibilizer) to 45/55–65/35 (with PS-graft-PA6 as compatibilizer).39 The influence of inorganic particles on the morphology of blends has also been widely studied during the last decade. For immiscible polymer/polymer/filler ternary blends, the development of phase structures may be more complicated. It has been speculated that exfoliated clay platelets or well dispersed nano-particles surrounded by an immobilized bound layer of polymer may hinder the particle coalescence by acting as physical barriers.40–42 Kontopoulou et al. found that the presence of clay in the dispersed phase results in reduced capability for particle break-up and a higher tendency for coalescence, resulting in the appearance of irregular particles.43 After that, Kontopoulou et al. reported the effect of nanosilica (SiO2) on the morphology of co-continuous immiscible PP/POE blends. They found that, upon addition of SiO2 in the presence of maleated PP compatibilizer, a finer structure consisting of elongated POE particles dispersed within the PP phase could be obtained.44 Izaro et al. studied the effects of location at the interface of an organoclay on the morphology and mechanical properties of a maleated-PP/PA6 co-continuous blend. The ductility behavior suggests that there is a maximum amount of organoclay that can be located at the interface while retaining its ductile nature. Once above this amount, both an inorganic barrier and discontinuity appear, hindering the stress transmission through the interface and leading to fragility.45
Polypropylene (PP) is a widely used semi-crystalline thermoplastic in many fields due to its good insulating and processing properties, small dielectric constant, relatively low price, as well as good stress crack and chemical resistance.46,47 However, its application is limited to a certain extent by its flammability. In order to overcome this disadvantage, compounding with flame retardants was adopted to improve its flame retardance. Among them, metal hydroxides have been widely utilized as truly “environmentally-friendly” additive-type flame retardants for polyolefins due to their non-toxicity, excellent smoke-suppression, and favorable cost consideration.48,49 Unfortunately, the addition of the metal hydroxides into polymers to meet the flame-retardant requirement is always extremely high, and the addition of high amounts of inorganic fillers would unavoidably deteriorate the mechanical properties of the resulting materials. Elongation at break is especially affected due primarily to the poor interfacial adhesion between the two components.50–53 However, in certain fields of industrial application, such as the material selection for wire and cable use, 10–15 MPa tensile strength and elongation at break larger than 150% is basically prerequisite. Therefore, a great deal of efforts has been made to solve this problem; unfortunately no perfect solution has yet been found. Generally, various elastomers are often used as the additional component to improve the toughness and flexibility of PP matrix, particularly for elongation at break. Among them, ethylene–octene copolymer (POE) has attracted more and more interest because it is highly elastomeric and tolerates high filler loadings while retaining its flexible properties.54–57 Moreover, POE has good compatibility with PP, is easily processed and recycled, which make it an ideal toughening modifier.58–61 Yin et al. investigated the fracture behaviour and deformation mechanism of PP/POE at presence of magnesium hydroxide (Mg(OH)2, MDH).55 The author presented three possible types of microstructures existing in such composites. They found that the average size of POE domains increased with increasing POE content until it reached a certain value, i.e. 80/20 PP/POE, after that the average size of the POE particles remained constant. Unfortunately, whether the co-continuous structure of PP/POE formed was not included. Moreover, little attention has been paid to the phase reversion of the polymer matrix, nor its effect on the mechanical properties of highly filled composites. Hong et al. prepared a series of flame retardant PP composites and investigated the effect of filler surface treatment and the effect of functionalized POE incorporation into PP matrix on their tensile properties as well as stress whitening during bending deformation.56 It was found that MDH coated with polymeric material give a high elongation at break value compared with the values obtained with uncoated and vinylsilane- or aminosilane-coated MDH. Besides, maleic anhydride grafted POE resulted in no stress whitening of the PP/POE/MDH composites, while neat POE led to whitening after compounding. However, again, no further investigations of the effect of co-continuous phase structure on the mechanical properties were carried out.
In this study, POE and MDH have been employed to modify PP simultaneously. We focus on co-continuous PP/POE blends and investigate the effect of high loading fillers on the morphology and mechanical properties of the composites. To our best knowledge, the influence of the high loading fillers on the co-continuous structure of the blends was not investigated yet. The relationships between phase structure and mechanical properties of PP/POE/MDH composites are discussed in detail.
Experimental
Materials
PP (1300, Yanshan Petrochemical, Beijing, China), with a melt flow rate (MFR) of 1.5 g/10 min (ASTM D 1238) and a density of 0.910 g cm−3, was used as the matrix polymer. POE (EXACTTM 5061, ExxonMobil Chemical, Houston, TX, USA) with a MFR of 0.9 g/10 min (ExxonMobil Method) and a density of 0.868 g cm−3 was selected as the modifier. The hydrophobic MDH powder was treated with coupling agent stearic acid before use, with an average particle size of 30 μm.
Sample preparation
The melt blending of the PP/POE blends containing 0–100 wt% POE was conducted in a twin-screw extruder (SLJ230, Sichuan Changlong Chemical Mechanical Equipment Company, Chengdu, China). During the extrusion, the screw speed was set at 130 rpm and the temperatures of various zones ranged from 180 to 200 °C. MDH was added to the PP/POE blends at loadings 50, 55 and 60 wt% respectively.
The testing samples of neat PP, neat POE, PP/POE blends and PP/POE/MDH composites for dynamic mechanical thermal analysis (DMTA) and mechanical analysis were prepared by compression molding. Pellets were preheated for 5 min at 200 °C and were then compressed for 3 min under a pressure of 10 MPa at 200 °C. After the pressure was released, the molded samples were removed from the press and cooled down to room temperature on a tabletop at STP.
Mechanical properties measurement
Tensile strength and elongation at break, as well as the yield strength of the samples were measured with a universal testing machine (CMT6503, Shenzhen SANS Test Machine Co., Ltd, Shenzhen, China) according to ASTM D882. The crosshead speed was set at 200 mm min−1. The tests were carried out in five folds.
Scanning electron microscopy (SEM) experiments
The morphology of the blends was studied by SEM after preferential etching of the POE phase in n-heptane for 2 h at 80 °C. The samples were cryogenically fractured in the direction perpendicular to the flow direction in liquid nitrogen before etching. The etched samples were carefully washed several times by using fresh n-heptane, and then washed three times with ethanol to remove the solvent. The samples were dried under vacuum at room temperature for 24 h to avoid any residual solvent. Then the fractured samples were observed in a SEM instrument (Inspect F, FEI Company, Hillsboro, OR, USA), using an acceleration voltage of 20 kV.
Dynamic mechanical thermal analysis (DMTA)
DMTA was performed with a dynamic mechanical thermo-analyzer (Q800, TA Instruments, New Castle, DE, USA) by using the single cantilever mode with a frequency of 1 Hz. The scanning temperature ranged from −95 to 150 °C, at a heating rate of 5 °C min−1 under a nitrogen atmosphere. The sample size was 11–13 mm in length, 6 mm in width, and 1 mm in thickness. The samples were heated before testing at 80 °C in a vacuum oven for 12 h to relieve any internal stress from molding.
Solvent extraction
In this manuscript, co-continuity in the blends were determined by extraction experiments. The preferential dissolution of the POE phase was performed in n-heptane for 2 h at 80 °C. After that, the samples were dried under vacuum at room temperature for 24 h. The tests were carried out in five folds to obtain an average value. In the case of co-continuity, 100% of the POE phase in PP/POE blends could be dissolved. Because it was not possible to find a solvent to extract the PP phase from PP/POE blends without damaging the remaining phase, the technique was only performed for the POE component. This parameter is calculated by the following equation:| |
 | (1) |
where w(POE) is the weight fraction of POE in the blend.
Results and discussion
Unfilled PP/POE blends
To better understand the properties and morphologies of the ternary composites, research has begun with PP/POE binary blends. Fig. 1 shows the tensile strength and elongation at break of the blends as a function of POE content. Compared to the neat PP, it is found that the tensile strength decreases about 7–45%, as the concentration of POE ranges from 10 wt% to 50 wt%. Specifically, the tensile strength slightly decreases from 33.6 MPa for pure PP to 31.2 MPa with 10 wt% of PP substituted by POE, then drops to 18.2 MPa for the blends as the POE content is increased to 30 wt%. Unexpectedly, for PP/POE blends with 50 wt% of POE, the tensile strength dramatically increased to 29.7 MPa. With further increase in the POE content to 70 wt% and 90 wt%, however, it drops again to 20.9 and 12.5 MPa, respectively. It is interesting to note that the blend at 50 wt% of POE content has an unordinary increased tensile strength, which may be explained by examining the morphology of the blends. Moreover, a rapid increase in elongation at break is achieved with POE addition rising from about 70% for pure PP to 250% for the blends at 10 wt%. The trend significantly increases to more than 1000% with further increased POE content above 30 wt%. The stress–strain curves of various PP/POE blends with different compositions are shown in Fig. 2. One observes that the yield strength, quite different from the tensile strength, decreases gradually with increasing POE content in the composition range studied. As for the blend with POE content below 30 wt%, there is obvious necking and the tensile strength is equal to the yield strength. Necking expresses the occurrence of a mechanical instability,62 so it does not lead to a stable new equilibrium and the sample approaches the point of fracture sooner. However, when POE content is equal or larger than 50 wt%, necking no longer exists and yield behavior is followed by strain hardening (an increase in stress with continued straining). Consequently, the tensile strength is much larger than the yield strength. One also observes that the stress–strain curves of 70/30 and 50/50 blends intersect at 800% strain, after that, the stress of the latter exceed that of the former. This indicates that the morphology in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 blends gives a synergistic effect on tensile strength.
:50 blends gives a synergistic effect on tensile strength.
 |
| | Fig. 1 Tensile properties of PP/POE blends versus POE content. | |
 |
| | Fig. 2 The stress–strain curves of various PP/POE blends with different POE content. | |
Fig. 3 shows the phase morphology of the samples after selectively etching of POE phase. The co-continuity data according to eqn (1) is shown in Table 1. When the POE content is 10 and 30 wt%, the SEM analysis of the blends (Fig. 3(a) and (b)) reveals that the minor component (POE) forms the dispersed phase and the major component (PP) forms the continuous phase as matrix. For the blends at 10 wt% of POE content, the sphere-like POE phase is uniformly dispersed in the PP matrix and the diameter of the domains never exceeds 1 μm. In the presence of such finely dispersed spherulite-like POE domains, the tensile strength remains unchanged. As the concentration of POE is increased further to 30 wt%, however, coalescence occurs and the dispersed phase transforms from a spherical domain to an elongated structure. The larger dispersed phase could very much reduce the specific surface and destroy the integrity of the matrix phase, which can act as a barrier for stress propagation. Consequently, the tensile strength is obviously deteriorated. As inferred from Fig. 3(c) and Table 1, at 50 wt% POE content, a fully co-continuous structure with an interpenetrating network is obtained. This is consistent with the observation of Jiao61 and Lee.44 In this case, the POE phase acts no longer as voids in the matrix, but as a fully co-continuous phase in the PP matrix. In other words, all of the POE component becomes part of the single percolating structure. This morphology combines the relatively higher rigidity PP and the higher flexibility POE. As for the POE phase, the PP crystallites can act as fillers and as cross-links which tends to increase the tensile strength of the POE component. In addition, the flexible POE component catalyzes the anisotropic orientation of the amorphous chains and the crystallites of PP that helps to realize a stable new equilibrium.54 This also contributes to the enhancement of the tensile strength of the blends. Therefore, thanks to the interpenetrating phase structure, both the elongation and the tensile strength of the blend are increased.
 |
| | Fig. 3 SEM micrographs of the PP/POE blends of different weight ratio of PP/POE: (a) 90/10; (b) 70/30; (c) 50/50. | |
Table 1 Continuity of the POE phase in the PP/POE blends
| PP/POE |
90/10 |
80/20 |
70/30 |
60/40 |
50/50 |
| Continuity (%) |
23.2 |
48.6 |
59.5 |
79.8 |
102.5 |
Influence of high-loading MDH on the co-continuous morphology of PP/POE blends
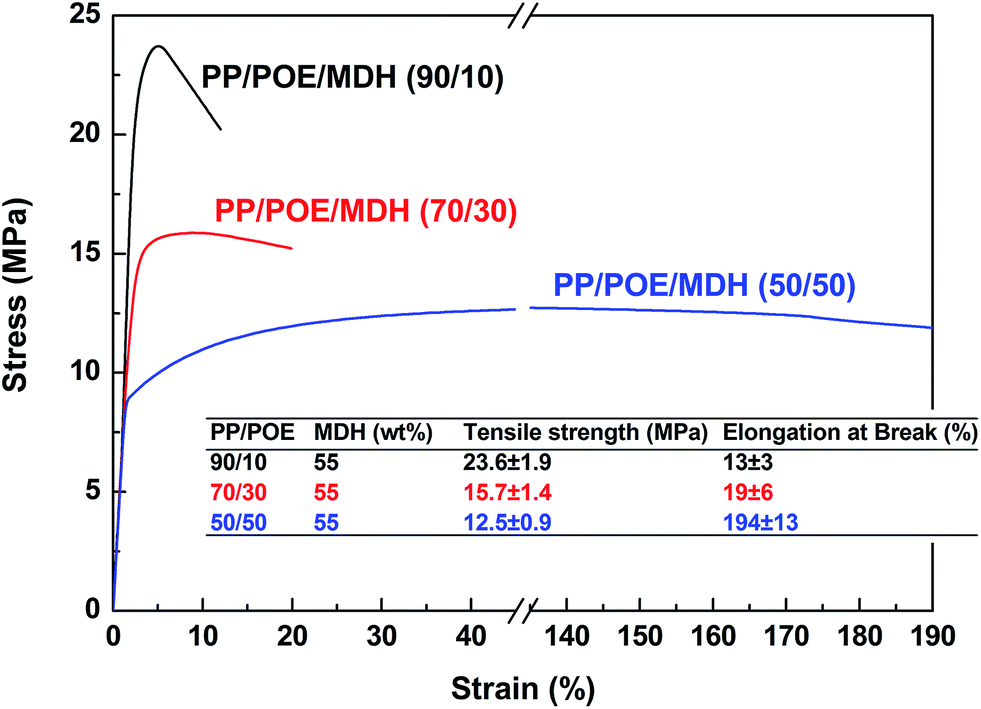
55 wt% of MDH has been added into the PP/POE blends, in order to investigate the influence of high-loading inorganic additives on the phase morphology of PP/POE blends, and thus the corresponding mechanical properties of the PP/POE/MDH ternary composites. PP/POE ratio is set at 90/10, 70/30 and 50/50 by weight respectively. Fig. 4 shows the stress–strain curves of different PP/POE/MDH composites. It can be found that the elongation at break of all samples show a sharp decrease after filled with 55 wt% of MDH. For the 90/10 and 70/30 blends, their elongations decrease from 250 and 1000% to the unacceptable levels of 16 and 19%, respectively, indicating that there is almost no plastic deformation before break. The deterioration is most likely due to the weak dispersion and aggregation of MDH, which heavily destroys the continuity of the PP matrix. For the 50/50 blends, however, the tensile ductility is obviously higher than that of the other two blends. It retains the relative high elongation at break near 200% after the addition of 55 wt% of MDH. This sudden increase in elongation is believed to be linked with the transformation of phase structure, which will be confirmed by other characterizations later in this study. On the other hand, it can be found that, for the highly filled composites, the tensile strength decreases gradually as the PP/POE ratio decreases. But even at the 50/50 PP/POE ratio, the tensile strength is still larger than 10 MPa. This indicates that acceptable mechanical properties can be achieved for halogen free flame retardant (HFFR) materials even at high loading of inorganic fillers, such as MDH and ATH (aluminium trihydroxide).63
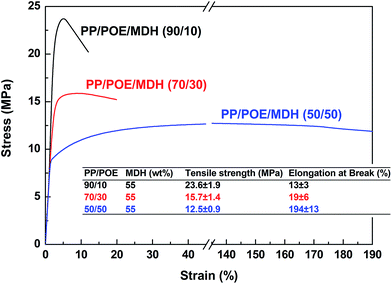 |
| | Fig. 4 The stress–strain curves of PP/POE/MDH composites with MDH content of 55 wt%. | |
SEM was carried out to understand the filler dispersion and its effect on the phase morphology of the resulting composites. Fig. 5 shows the phase morphology of the samples after selectively etching of the POE phase. For PP/POE/MDH composites with PP/POE ratio of 90/10 or 70/30, both the filler and POE are separately dispersed in the PP matrix with the MDH particles size of 0.3–10 μm. In these two cases, the boundaries and the contrast between the MDH filler and the PP matrix can be clearly observed. It can also be found that part of the smaller particles begin to concentrate and aggregate in certain locations, which causes the deterioration of the mechanical properties of composites; also the poor adhesion between the filler and matrix further decrease the mechanical properties. As compared with the corresponding unfilled blends, the number of and the average diameter of the POE domains decreases upon the addition of high loading MDH. Moreover, the elongated structure of POE domain, particularly in the 70/30 blend, no longer exists in the highly filled composite with the same PP/POE weight ratio. This is mostly because a great amount of rigid MDH fillers facilitate the breaking up of POE domains and inhibit their further aggregation. Also, since the PP and POE phase have the same probability to encapsulate the MDH fillers, part of the POE component is consumed to wet the MDH surface. In other words, the effect of adding high loading fillers, in this respect, is equal to lowering the POE content. However, for PP/POE/MDH composite with a PP/POE ratio of 50/50, an evident co-continuous phase structure is still formed (the continuity of POE phase is unavailable because part of MDH particles are also removed with the extraction of POE phase). This indicates that the co-continuous morphology can be achieved at 50/50 PP/POE ratio and maintained even at high loading of fillers.
 |
| | Fig. 5 SEM micrographs of the PP/POE/MDH composites (55 wt% of MDH) of different weight ratio of PP/POE: (a) 90/10; (b) 70/30; (c) 50/50. | |
Influence of filler content on co-continuous morphology
The morphology of the PP/POE/MDH ternary composites with different filler content was also investigated, as shown in Fig. 6. This time, the PP/POE ratio by weight remains at 50/50, while the filler content varies from 50 wt% to 60 wt%. It can be seen that the agglomeration of the MDH fillers becomes more obvious with the increase of MDH content while the co-continuous structure remains almost unchanged. When MDH content is up to 60 wt%, a large amount of fillers aggregate together, but the co-continuous structure of PP and POE can still be clearly observed. This result indicates that the filler content hardly influences the co-continuous structures of the polymer composite. It was reported that the composition of the two polymers required to form the co-continuous structure is mainly determined by the viscosity ratio and interfacial tension between the two components.11,13 According to Favis et al.'s theory, moreover, the formation of the co-continuous structure in a binary blend is due to the coalescence of the minor phase domains.29 In our cases, inorganic filler is randomly dispersed in both PP and POE phases, suggesting that its effect on viscosity ratio is negligible and the suppressing effect on the calescence of both polymer phases is similar. Therefore, the morphological changes are attributed to the compositional effect of the polymer composite and are independent of the filler content.
 |
| | Fig. 6 SEM micrographs of the PP/POE/MDH composites (weight ratio of PP/POE as 50/50) with different MDH contents: (a) 50 wt%; (b) 55 wt%; (c) 60 wt%. | |
Dynamic mechanical thermal analysis (DMTA) of the composites
Dynamic mechanical thermal analysis (DMTA) is a technique widely used to study and characterize materials' viscoelastic behavior, glass transition temperature, and thermo-mechanical transitions corresponding to other molecular motions. DMTA can also provide valuable information about the structure–property relationships of the systems. Fig. 7 shows the curves of tanδ of the unfilled PP/POE blends and highly filled PP/POE/MDH composites versus temperature, respectively. Two tanδ peaks correspond to the glass transitions of the POE and PP components in the samples and the corresponding data are shown in Table 2. The relative increment of peak value (ΔPr) is defined to indirectly confirm the effect of high loading fillers on the phase structure of the samples.64 It is deduced from the following expression:| |
 | (2) |
where Pfilled and Punfilled represent the tanδ peak value of filled composite and unfilled blend, respectively. It is well known that the tanδ value is related to the damping performance of the material;65 while in this case, addition of the high loading fillers restricts the mobility of the polymer chains and hence lowers the damping performance of both the PP and POE components. Therefore, all samples highly filled with MDH show decreased tanδ peak values along with a Tg shift to higher temperatures. For 90/10 and 70/30 systems, the POE domains and MDH fillers are separately dispersed in the PP matrix, although the average diameter of the POE domain is much smaller than that of the MDH filler (Fig. 5(a) and (b)). In this case, a large proportion of the POE domains are shielded by the MDH fillers. Thus, less POE is able to be deformed during the DMTA test conditions reducing the amount of dissipated energy of POE. In this respect, a larger ΔPr of POE than that of PP is expected to be revealed in a tanδ versus temperature thermogram. In the 50/50 system, however, the ΔPr of POE is much lower than the values of other two systems, and it is approximately equal to that of PP, indicating that the shielding effect caused by the highly loaded fillers no longer exists. This can be explained by the dispersed POE domains agglomerating to become part of a single percolating phase (Fig. 4(c)), which confirms the formation of a co-continuous structure in this composite. On the other hand, once the temperature is higher than the glass transition temperature of the POE component, fillers dispersed in the continuous POE phase largely could enhance the intermolecular friction between POE phase and fillers during the DMTA test conditions, leading to positive impact on the damping performance of the samples, this explains why the ΔPr of PP in the co-continuous case is much lower than the other two cases.
 |
| | Fig. 7 tanδ of different PP/POE blends and PP/POE/MDH composites (55 wt% of MDH) as a function of temperature obtained by DMTA test: (a) 90/10; (b) 70/30; (c) 50/50. | |
Table 2 The glass transition temperature (Tg), peak value, and relative increment of peak value (ΔPr) of PP and POE in PP/POE binary blend and PP/POE/MDH ternary composite
| Sample |
PP/POE |
MDH (wt%) |
Tg (°C) |
Peak value |
ΔPr (%) |
| POE |
PP |
POE |
PP |
POE |
PP |
| 1 |
90/10 |
— |
−31.7 |
25.6 |
0.036 |
0.078 |
— |
— |
| 1′ |
90/10 |
55 |
−29.5 |
28.7 |
0.023 |
0.055 |
36.1 |
29.5 |
| 2 |
70/30 |
— |
−31.9 |
21.1 |
0.064 |
0.087 |
— |
— |
| 2′ |
70/30 |
55 |
−26.1 |
26.9 |
0.040 |
0.070 |
37.5 |
19.5 |
| 3 |
50/50 |
— |
−30.3 |
23.5 |
0.075 |
0.079 |
— |
— |
| 3′ |
50/50 |
55 |
−28.4 |
27.6 |
0.072 |
0.077 |
4 |
2.5 |
The storage modulus versus temperature plots for different polymer blends also give useful results to distinguish between dispersed and co-continuous structures. In dispersed structures, the storage modulus is mainly determined by the matrix component, whereas in co-continuous structures the storage modulus–temperature dependence reflects a greater contribution of both components.66–68 Therefore, the storage modulus would drastically change at the phase inversion region of the blends. Fig. 8 shows the storage modulus as a function of the temperature for different PP/POE/MDH composites. It is observed that the storage modulus increases with increasing POE content at the temperature below the glass transition temperature of POE about −25 °C. The difference is that the sample at PP/POE 50/50 demonstrates a more dramatic increase in the plateau region between −100 and −50 °C, this can be explained by the greater contribution of the POE phase to the storage modulus, which also confirms the formation of a co-continuous structure in this composite.
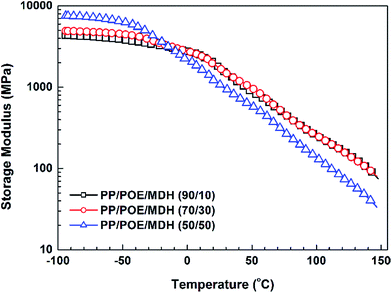 |
| | Fig. 8 Storage modulus of different PP/POE/MDH composites (55 wt% of MDH) as a function of temperature obtained by DMTA test: (a) 90/10; (b) 70/30, and (c) 50/50. | |
Deformation mechanism of PP/POE/MDH composites
Dubnilova et al. proposed that the tensile morphologies of the PP/Al(OH)3 composites are controlled by the relationship between the plastic deformation strength of the craze-like zones (σcz) (eqn (3)) and the debonding strength (σd).69 They revealed that the strain-harden phenomenon in PP/Al(OH)3 composites disappeared when the Al(OH)3 content exceeds 20 vol%.where σbh is the plastic deformation strength of polymer bulkhead, Φ is the filler volume content and α presents the fraction of the debonded filler particles. When σcz is larger than σd, the debonding process can extend to both sides of the samples in the loading direction, which leads to the ductile fracture; when σcz is less than σd, the debonded zones cannot be extended, which leads to the brittle fracture. For the PP/MDH composites, in our cases, the initial deformation is due to the debonding behavior of the MDH filler and the debonded region can't be extended, which leads to the brittle fracture. For the POE/MDH composites, because of the ductile property of POE, the initial deformation is the elastic deformation, after that the strain-hardening phenomenon occurs in the tensile process, which increases the deformation strength till larger than the debonding strength, then the plastic deformation and debonding process of the MDH filler from the POE matrix occurs. The strain-hardening effect is thought to be more useful for a filled composite designed for high elongation at break.56,70 For PP/POE/MDH ternary composites, although the deformation mechanism is more complicated because of the coexistence of PP and POE, it's not difficult to understand that the deformation mechanism is similar as PP/MDH composites when POE and MDH are separately dispersed in the PP matrix; while for the PP/POE/MDH composites when co-continuous structure is formed, its deformation mechanism falls between PP/MDH composites and POE/MDH composites. Therefore, the co-continuous structure is the key to achieve a higher elongation at break and acceptable tensile strength.
Conclusions
In this manuscript, co-continuous phase structures can be formed at 50/50 weight ratio of PP/POE both in PP/POE blend and PP/POE/MDH ternary composites, even if the content of MDH is high as 60 wt%. Upon the addition of 55 wt% MDH fillers, the droplet/matrix structures show smaller average dimensions of the POE domains while the co-continuous structure remains almost unchanged, as observed by SEM and further confirmed by the DMTA results. Furthermore, the filler content has no obvious effect on the co-continuous structure, even at ultra-high filling content. In the co-continuous case, the samples highly filled with MDH fillers show satisfying mechanical performances, maintaining both the tensile strength and elongation at break at acceptable levels for practical applications. The strain-hardening effect of the POE component in the tensile process is believed to be responsible for the higher elongation at break of the composites. This result indicates that the co-continuous structure is in favour of tolerating high loading fillers while retaining flexible properties.
Acknowledgements
Financial supports by the National Natural Science Foundation of China (grant no. 51121001) and Program for Changjiang Scholars and Innovative Research Team in University (IRT 1026) would be sincerely acknowledged.
Notes and references
- K. Prephet and P. Horanont, Polymer, 2000, 41, 9283–9290 CrossRef.
- A. Mamat, T. Vu Khanh, P. Cigana and B. D. Favis, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 2583 CrossRef CAS.
- U. Niebergall, J. Bohse, B. L. Schürmann, S. Seidler and W. Grellmann, Polym. Eng. Sci., 1999, 39, 1109 CAS.
- H. Veenstra, P. C. J. Verkooijen, B. J. J. van Lent, J. van Dam, A. P. de Boer and A. Nijhof, Polymer, 2000, 41, 1817 CrossRef CAS.
- S. Jouenne, J. A. Gonzalez-Leon, A. V. Ruzette, P. Lodefier and L. Leibler, Macromolecules, 2008, 41, 9823 CrossRef CAS.
- P. Potschke, A. R. Bhattacharyya and A. Janke, Polymer, 2003, 44, 8061 CrossRef CAS PubMed.
- J. M. Frost, F. Cheynis, S. M. Tuladhar and J. Nelson, Nano Lett., 2006, 6, 1674 CrossRef CAS PubMed.
- P. Pötschke and D. R. Paul, J. Macromol. Sci., Part C, 2003, 43, 87 CrossRef PubMed.
- F. Gubbels, S. Blacher, E. Vanlathem, R. Jérôme, R. Deltour and F. Brouers, Macromolecules, 1995, 28, 1559 CrossRef CAS.
- K. Cheah, M. Forsyth and G. P. Simon, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 3106 CrossRef CAS.
- R. C. Willemse, A. Speijer, A. E. Langeraar and A. P. de Boer, Polymer, 1999, 40, 6645 CrossRef CAS.
- P. Sarazin and B. D. Favis, Biomacromolecules, 2003, 4, 1669–1679 CrossRef CAS PubMed.
- R. C. Willemse, A. Posthuma de Boer, J. van Dam and A. D. Gotsis, Polymer, 1998, 39, 5879 CrossRef CAS.
- J. Lyngaae-JΦgensen, Int. Polym. Process., 1999, 14, 213–220 CrossRef.
- K. Dedecker and G. Groeninckx, Polymer, 1998, 39, 4993–5000 CrossRef CAS.
- D. Bourry and B. D. Favis, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 1889 CrossRef CAS.
- N. Marin and B. D. Favis, Polymer, 2002, 43, 4723 CrossRef CAS.
- J. A. Galloway, M. D. Montminy and C. W. Macosko, Polymer, 2002, 43, 4715 CrossRef CAS.
- S. Steinmann, W. Gronski and C. Friedrich, Polymer, 2001, 42, 6619 CrossRef CAS.
- J. A. Galloway and C. W. Macosko, Polym. Eng. Sci., 2004, 44, 714 CAS.
- A. Ajji, L. Choplin and R. E. Prudhomme, J. Polym. Sci., Part B: Polym. Phys., 1991, 29, 1573 CrossRef CAS.
- S. Mani, M. F. Malone and H. H. Winter, J. Rheol., 1992, 36, 1625 CrossRef CAS.
- S. Steinmann, W. Gronski and C. Friedrich, Polymer, 2002, 43, 4467 CrossRef CAS.
- C. E. Scott and C. W. Macosko, Polymer, 1995, 36, 461 CrossRef CAS.
- U. Sundararaj, Y. Dori and C. W. Macosko, Polymer, 1995, 36, 1957 CrossRef CAS.
- D. R. Paul and J. W. Barlow, J. Macromol. Sci., Part C, 1980, 18, 109 CrossRef.
- C. L. Zhang, L. F. Feng, J. Zhao, H. Huang, S. Hoppe and G. H. Hu, Polymer, 2008, 49, 3462 CrossRef CAS PubMed.
- J. M. Li, P. L. Ma and B. D. Favis, Macromolecules, 2002, 35, 2005 CrossRef CAS.
- A. Leclair and B. D. Favis, Polymer, 1996, 37, 4723 CrossRef CAS.
- Y. T. Hu, D. J. Pine and L. G. Leal, Phys. Fluids, 2000, 12, 484 CrossRef CAS PubMed.
- W. J. Milliken and L. G. Leal, J. Colloid Interface Sci., 1994, 166, 275 CrossRef CAS.
- H. A. Stone and L. G. Leal, J. Fluid Mech., 1990, 220, 161 CrossRef CAS.
- B. Majumdar, H. Keskkula and D. R. Paul, Polymer, 1994, 35, 3164 CrossRef CAS.
- N. Mekhilef, B. D. Favis and P. J. Carreau, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 293 CrossRef CAS.
- S. H. Jafari, P. Potschke, M. Stephan, G. Pompe, H. Warth and H. Alberts, J. Appl. Polym. Sci., 2002, 84, 2753 CrossRef CAS.
- F. S. Bates, W. W. Maurer, P. M. Lipic, M. A. Hillmyer, K. Almdal and K. Mortensen, Phys. Rev. Lett., 1997, 79, 849 CrossRef CAS.
- M. A. Hillmyer, W. W. Maurer, T. P. Lodge, F. S. Bates and K. Almdal, J. Phys. Chem. B, 1999, 103, 4814 CrossRef CAS.
- J. M. Willis, V. Caldas and B. D. Favis, J. Mater. Sci., 1991, 26, 4742 CrossRef CAS.
- C. L. Zhang, L. F. Feng, J. Zhao, H. Huang, S. Hoppe and G. H. Hu, Polymer, 2008, 49, 3462 CrossRef CAS PubMed.
- H. Lee, P. D. Fasulo, W. R. Rodgers and D. R. Paul, Polymer, 2005, 46, 11673 CrossRef CAS PubMed.
- B. B. Khatua, D. J. Lee, H. Y. Kim and J. K. Kim, Macromolecules, 2004, 37, 2454 CrossRef CAS.
- J. R. Austin and M. Kontopoulou, Polym. Eng. Sci., 2006, 46, 1491 CAS.
- M. Kontopoulou, Y. Liu, J. R. Austin and J. S. Parent, Polymer, 2007, 48, 4520 CrossRef CAS PubMed.
- S. H. Lee and M. Kontopoulou, Polymer, 2010, 51, 1147 CrossRef CAS PubMed.
- I. Goitisolo, I. González and J. I. Eguiazábal, Polym. Adv. Technol., 2013, 24, 357 CrossRef CAS.
- H. W. Bai, Y. Wang, B. Song and L. Han, J. Appl. Polym. Sci., 2008, 108, 3270 CrossRef CAS.
- Q. Zheng, Y. G. Shangguan, S. K. Yan, Y. H. Song, M. Peng and Q. B. Zhang, Polymer, 2005, 46, 3163 CrossRef CAS PubMed.
- F. Laoutid, L. Bonnaud, M. Alexandre, J. M. Lopez-Cuesta and P. Dubois, Mater. Sci. Eng., R, 2009, 63, 100 CrossRef PubMed.
- L. Chen and Y. Z. Wang, Polym. Adv. Technol., 2010, 21, 1 Search PubMed.
- G. L. Wang, P. K. Jiang, Z. K. Zhu and J. Yin, J. Appl. Polym. Sci., 2002, 86, 2544 CrossRef CAS.
- A. Silva, M. Rocha, F. Countinho, M. Rocha, F. Countinho, R. Bretas and C. Scuracchio, J. Appl. Polym. Sci., 2000, 75, 692 CrossRef.
- C. M. Tai and R. K. Y. Li, J. Appl. Polym. Sci., 2001, 80, 2718 CrossRef CAS.
- G. L. Wang, P. K. Jiang, Z. K. Zhu and J. Ying, J. Appl. Polym. Sci., 2002, 85, 2485 CrossRef CAS.
- Y. F. Men and J. Rieger, Phys. Rev. Lett., 2003, 91, 095502 CrossRef.
- J. Yin, Y. Zhang and Y. X. Zhang, J. Appl. Polym. Sci., 2005, 98, 957 CrossRef CAS.
- C. H. Hong, Y. B. Lee, J. W. Bae and J. Y. Jho, J. Appl. Polym. Sci., 2005, 97, 2311 CrossRef CAS.
- H. Shen, Y. H. Wang and K. C. Mai, Thermochim. Acta, 2009, 483, 36 CrossRef CAS PubMed.
- T. McNally and P. McShane, Polymer, 2002, 43, 3785 CrossRef CAS.
- N. Zeng, S. L. Bai, C. G'Sell, J. M. Hiver and Y. W. Mai, Polym. Int., 2002, 51, 1439 CrossRef CAS.
- X. W. Liu, L. J. Ji and Z. Wang, China Plast. Ind., 2008, 36, 29 Search PubMed.
- Y. H. Jiao, X. L. Wang, Y. Z. Wang, L. Chen, S. M. Mu and F. Li, J. Macromol. Sci., Part B: Phys., 2009, 48, 351 CrossRef CAS.
- P. Vincent, Polymer, 1960, 1, 7 CrossRef CAS.
- L. Yu, L. Chen, L. P. Dong, L. J. Li and Y. Z. Wang, RSC Adv., 2014, 4, 17812 RSC.
- H. Yang and Q. Fu, Polymer, 2007, 48, 860–869 CrossRef CAS PubMed.
- A. P. Laly, O. Zachariah and T. Sabu, Compos. Sci. Technol., 2003, 63, 283 CrossRef.
- K. Dedecker and G. Groeninckx, Polymer, 1998, 39, 4993 CrossRef CAS.
- D. Quintens, G. Groeninckx, M. Guest and L. Aerts, Polym. Eng. Sci., 1990, 30, 1474 CAS.
- P. Potschke and D. R. Paul, Macromol. Symp., 2003, 198, 69 CrossRef.
- I. L. Dubnikova, D. K. Muravin and V. G. Oshmyan, Polym. Eng. Sci., 1997, 37, 1301 CAS.
- K. Sirisinha and W. Kamphunthong, Polym. Test., 2009, 28, 636 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.