
Mechanism evolution for the oxidative dehydrogenation of ethyl benzene to styrene over V2O5/TiO2 catalyst: computational and kinetic approach†
Abstract
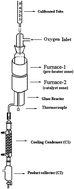
A kinetic study of the oxidative dehydrogenation of ethylbenzene has been performed in a differential flow reactor over a V2O5/TiO2 catalyst. The partial pressures of ethylbenzene and oxygen were varied and rates were measured for the formation of styrene. Kinetics studies reveal that the mechanism of the reaction is of the “Mars Van Krevelen Redox” type. The rate equation deduced, assuming a steady state involving a two stage oxidation–reduction process, represented the data most satisfactorily. The catalyst has been synthesized by a method similar to a solution combustion method and characterized using FT-Raman and powder XRD methods. DFT level computational studies provided a deep insight for the reaction mechanism at the molecular level. According to our calculations ethylbenzene first dissociatively adsorbs over the catalyst and produces an ethylbenzene radical which subsequently adsorbs on the vanadyl group or bridging oxygen V–O–V or bridging oxygen V–O–Ti. The adsorbed radical further loses an H atom to produce styrene. The catalyst is regenerated by molecular oxygen.
 Please wait while we load your content...
Please wait while we load your content...

