
Ordered short channel mesoporous silica modified with 1,3,5-triazine–piperazine as a versatile recyclable basic catalyst for cross-aldol, Knoevenagel and conjugate addition reactions with isatins†
Naveen Guptaab,
Tamal Roya,
Debashis Ghoshab,
Sayed H. R. Abdi*ab,
Rukhsana I. Kureshyab,
Noor-ul H. Khanab and
Hari C. Bajajab
aDiscipline of Inorganic Materials and Catalysis, CSIR-Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), G. B. Marg, Bhavnagar-364 002, Gujarat, India. E-mail: shrabdi@csmcri.org; Fax: +91 0278 2566970
bAcademy of Scientific and Innovative Research, CSIR-Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), G. B. Marg, Bhavnagar-364 002, Gujarat, India
First published on 5th February 2015
Abstract
A triazine–piperazine immobilized on ordered short channel mesoporous silica was synthesized, characterized and used as a heterogeneous basic catalyst in the synthesis of indole skeletal structures from isatin derivatives under ambient reaction conditions in good to excellent isolated yields. The catalyst showed no loss in its efficacy over 10 recycle experiments.
Introduction
The search for amenable synthetic routes to synthesize natural products (alkaloids) and pharmacologically active agents bearing functionalized indole moieties has always been on the agenda of synthetic chemists over the decades.1,2 With increasing developments in synthetic chemistry, the possibility of finding newer molecules containing the indole moiety as drug candidates mimicking active natural products has increased many folds.3 For the synthesis of a functionalized indole nucleus, the use of isatin and its derivatives as starting motif have received particular attention owing to their straight forward synthesis and commercial availability.4 A range of organic transformations such as aldol condensation,5 Knoevenagel condensation,6 conjugate addition,7 and allylation8 have thus far been reported where isatin and their derivatives were utilized as starting materials for the synthesis of targeted indoles. In general, these reactions involve nucleophilic attack at the β-position (or 3-position) of the isatin moiety in the presence or even absence of a catalyst to produce a wide variety of indole derivatives.9 Organocatalysts have been particularly favoured for this purpose which are mainly developed by mimicking the enzymes known to catalyze such reactions.10 Despite the significant success in nucleophilic substitution reactions of isatins, there is still sufficient room for improvement, particularly in terms of catalyst loading and its recovery and reuse.11Typically for aldol condensation,12 Knoevenagel reaction,13 Michael addition14 and conjugate addition15 reactions, secondary amines derived from homogeneous as well as heterogeneous catalysts have been employed. Piperazine, in particular, has shown interesting properties as an organocatalyst in nucleophilic substitution reactions of carbonyl compounds owing to its ability to form enamine, which is thought to be the reactive intermediate for aldol condensation.16 Very recently M. Belén-Cid and co-workers have reported piperazine bound reduced graphene oxide as a reusable catalyst for Knoevenagel, Michael and aldol reactions for simple aldehydes.17 Yuan et al.18 in 2009 demonstrated that cross-aldol condensation of isatins with ketones can be achieved in the absence of a catalyst. However this protocol works only under strict anhydrous condition and in the presence of DMF as a solvent, whose role is not well understood. With this backdrop, we decided to explore various secondary amines as basic catalysts for obtaining indole derivatives from isatins. Among the secondary amines used, piperazine and its tri-piperazine–triazine form showed higher catalytic activity under homogenous condition, which formed the basis for considering silica–triazine–piperazine hybrid (STP-hybrid) as the heterogeneous catalyst for the present study. Incidentally, our group has used STP-hybrid as a core to build catalysts for asymmetric epoxidation reaction.19 In order to overcome the diffusional constrains often associated with such silica materials, the silica used in this case was short channel periodic mesoporous silica. With the use of STP-hybrid as a catalyst for the present organic transformations we got excellent isolated yield (up to 98%) and product selectivity (up to ≥99%). In addition to this the STP-hybrid catalyst was successfully recycled for 10 recycle experiments studied.
Results and discussion
In quest for the synthesis of substituted indole derivatives, the cross-aldol of acetone (also as solvent in these cases) to isatin (Table 1) was performed in the presence of three representative secondary amines as organocatalysts viz. piperazine (Table 1, entry 1), pyrolidine (Table 1, entry 2) and di-isopropylamine (Table 1, entry 3) at room temperature (RT). Among these, piperazine showed the highest reactivity (Table 1, entry 1).
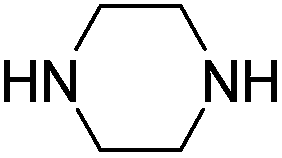


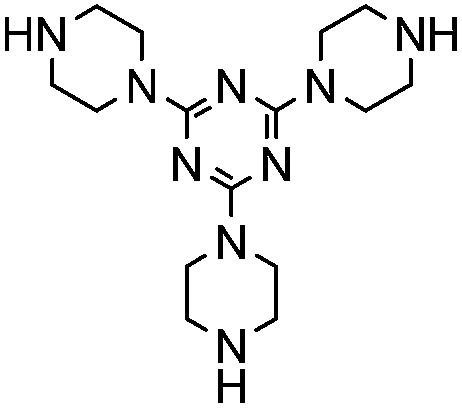
To further improve our results and in order to make the catalyst recyclable, we increased the molecular weight of the catalyst by incorporating piperazine on 1,3,5-triazine ring and used it for this reaction under similar reaction condition. Although we got better reactivity (Table 1, entry 4) as the reaction got completed one hour earlier than in the case of piperazine, but this catalyst was soluble in the reaction medium and required chromatographic separation in the post catalytic work-up. To deal this issue, we thought of heterogenizing piperazine by supporting it on silica. For this purpose we took a clue from the work recently reported by our group on melamine–piperazine core supported on short channel ordered mesoporous silica for immobilization of Mn(III) salen complexes in asymmetric epoxidation of non-functionalized olefins.19 We envisioned that the above mentioned supported melamine–piperazine core might work well in the present scenario too. We speculated the following many fold advantages for this strategy over simple piperazine catalyst immobilized on solid inorganic supports: (i) large pore size and short channel length of the support material would minimize the diffusional constrains for the reactants and product, (ii) the presence of a melamine linker would increase the effective nitrogen content which is believed to be beneficial for base catalyzed organic transformation as it increases the effective basicity of the catalyst,20 (iii) presence of a longer linker would keep the catalytically active piperazine site at a larger distance from the support material which would prevent any negative electronic or steric effect of the support material on the catalytic activity and lastly (iv) the presence of more than one catalytically active sites may in turn reduce the catalyst loading due to the possible tandem interaction between two catalytic centers.
Accordingly, we synthesized the heterogeneous dimeric piperazine catalyst (IMPC) grown on a melamine core following the reported21 procedure (Scheme 1). The support-AFMS and the immobilized catalyst (IMPC) were characterized by appropriate physico-chemical techniques such as diffuse reflectance spectra, IR, solid state NMR, SEM, TEM, XRD, surface area measurements and thermo gravimetric analysis.
 | ||
| Scheme 1 Synthesis of immobilized catalyst on mesoporous silica support. | ||
Characterization of the support and the catalyst
The N2 adsorption–desorption isotherm of the support material provided in Fig. 1 exhibited typical type IV isotherms akin to the mesoporous material with narrow pore size distribution centered around 5.4 nm similar to that of periodic mesoporous silica. | ||
| Fig. 1 N2 adsorption and desorption isotherm and pore width profile of (AFMS). | ||
Upon functionalization with the melamine–piperazine moiety, both the surface area and the pore size decreased (Table 2), but the nature of the isotherm remained same. This clearly indicates the successful immobilization of the melamine piperazine catalyst without disturbing the periodic nature of the support.
| Compound | Total pore volume (cm3 g−1) | BJH pore width (Å) | BET surface area (m2 g−1) |
|---|---|---|---|
| a Textural parameters of AFMS and IMPC. | |||
| AFMS | 1.14 | 66.97 | 628 |
| IMPC | 0.97 | 66.13 | 556 |
The powder X-ray diffraction (PXRD) pattern of the support material-AFMS showed corresponding peak to the reflections along 100, 110 and 200 planes justifying a uniform hexagonal lattice structures much similar to that observed in the case of SBA-15.22 After functionalization with the melamine–piperazine core, the basic PXRD pattern remained the same, however a decrease in peak intensity and minor shift to the lower 2θ value were observed (in line with our previous report),19 which further supports successful loading of the melamine–piperazine core on AFMS (see ESI†).
The SEM images of the AFMS showed the hexagonal disc-like morphology (disc size (1 to <2 μm)), majority of which were nicely stacked on each other (Fig. 2).
 | ||
| Fig. 2 SEM images of support material AFMS (a) and immobilized catalyst IMPC (b). | ||
The TEM images of the AFMS, when viewed along the short axis, showed the presence of hexagonal pore structure with well-defined walls, while from side view equidistant parallel fringes could be seen, which was in consonance with PXRD pattern (Fig. 3). Both the SEM and TEM pattern remained intact after immobilization of melamine–piperazine core, thereby confirms the robust mechanical, thermal and chemical stability of the AFMS.
 | ||
| Fig. 3 TEM images of support material AFMS (a) and immobilized catalyst IMPC (b). | ||
The loading of the effective homogeneous melamine–piperazine unit on the support material was found to be 17 mg/100 mg as determined from TGA analysis (see ESI† for TGA profile).
Basicity of the support material AFMS and immobilized catalyst IMPC was determined by Hammett indicator experiment (for details see ESI†).
FT-IR spectra of the ordered mesoporous silica support showed the characteristic bands at 2979, 1640, 1079 cm−1 corresponding to the C–H stretching frequency of the amino-propyl arm of the silylating agent, the bending vibration for the silanol O–H groups and asymmetric Si–O–Si stretching respectively.
Upon functionalization, the new bands at 1544 (1,3,5 s-triazing ring “quadrant stretch”) and 1449 (1,3,5 s-triazing ring “semicircle stretch”)23 supports the successful immobilization of the melamine unit on mesoporous silica support. However, the presence of the piperazine unit couldn't be confirmed on the basis of IR which is perhaps due to overlap of its characteristic peaks with that of the amino-propyl unit of silylating agent (Fig. 4).
 | ||
| Fig. 4 IR spectra of support material AFMS and immobilized catalyst IMPC. | ||
Fig. 5 represents the UV-Vis diffuse reflectance spectra of the mesoporous solid support material which did not show any distinct band in the entire region. However, the immobilized catalyst showed two new bands at around 241 nm and 336 nm assigned to π–π* and n–π* transitions originating from 1,3,5-triazine unit of melamine–piperazine core.
 | ||
| Fig. 5 UV-Vis DRS spectra of support material AFMS and immobilized catalyst IMPC. | ||
Solid state NMR spectra
The solid state 13C{1H} CP-MAS spectra of IMPC showed peaks at 166.7 (aromatic C of 1,3,5-triazine), 44.3 (aliphatic C–N of piperazine merged with C–N of amino propyl arm), 23.1 (CH2 of amino propyl arm) and 10.6 (Si–CH2 of amino propyl arm) ppm which confirms the presence of melamine–piperazine core in its structure (see ESI†).All the characterization data presented earlier together confirm the successful immobilization of melamine–piperazine catalyst (IMPC) on mesoporous silica support keeping intact its structural properties.
Activity of piperazine-immobilized silica IMPC in cross-aldol reaction of isatins
Once characterized, the piperazine-immobilized silica IMPC was employed as catalyst in aldol addition of acetone (1a) (both as nucleophile and solvent) to isatin (2a) Scheme 2. | ||
| Scheme 2 Solvent and catalyst optimization. | ||
In the search of optimized reaction condition, initially 50 mg of IMPC (corresponds to 5 mol% of piperazine) was taken together with isatin (0.3 mmol) and acetone (1 mL), which gave complete conversion of the isatin to yield aldol adduct 3aa in 6 h at room temperature (Fig. 6a, entry 2). An increase in catalyst loading to 100 mg (Fig. 6a, entry 1) made the reaction slightly faster, whereas a decrease in catalyst loading to 25 mg (Fig. 6a, entry 3) resulted only marginal increase in reaction time (Fig. 6a). However, on further decreasing the amount of the solid catalyst to 10 mg, the rate of the reaction dropped significantly (Fig. 6a, entry 4), which prompted us to choose 25 mg loading of the solid catalyst as optimum (entry 3). Noticeably, in none of the catalytic experiments self-condensation products were observed (based on 1H-NMR of the crude products).
 | ||
| Fig. 6 (a) Optimization of catalyst loading. (b) Screening of solvent. | ||
Once we had the optimized catalyst loading data at our hand, the reaction medium (Fig. 6b) was varied keeping acetone (10 equivalent) as the source of nucleophile. Any attempt to reduce the quantity of acetone resulted in significant increase in reaction time. Among all the solvents studied, THF was found to be the best solvent for this reaction; nevertheless even THF could not match the performance of the catalyst when neat acetone was used both as solvent as well as a reactant (Fig. 6a, entry 3).
After optimizing the reaction condition, we further investigated the substrate scope in aldol addition of various ketones (1a–1f) to isatin and its derivatives (2a–2l). The data listed in Table 3 showed that in almost all the cases the reaction proceeded smoothly to afford the aldol products (3aa–3hf). However, the aldol reaction was significantly slower for the substituted isatins as compared to virgin isatin, irrespective of the electronic nature of the substituent on isatin core. Strikingly, the addition of acetone to 5-fluoro isatin, gave the corresponding aldol adduct at a much faster rate as compared to their chloro or bromo counterparts. Furthermore, for different bromo substituent isatins, the order of activity was found to be 5-substituted > 6-substituted > 4-substituted.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | R3 | Solvent | Time [h/days] | Yieldb [%] |
| a Reaction condition: isatins (0.3 mmol), nucleophile (acetone-1 mL or 5 eq. ketone derivatives) THF (1 mL), catalyst (25/50 mg), RT.b Isolated yield.c Catalyst loading: 25 mg.d Catalyst loading: 50 mg. | |||||||
| 1c | 2a | CH3 (1a) | H | H | Neat | 6 h | > 98 |
| 2c | 2b | CH3 (1a) | H | Me | Neat | 20 h | 90 |
| 3c | 2c | CH3 (1a) | H | Ph | Neat | 30 h | 92 |
| 4c | 2d | CH3 (1a) | 7-Cl | H | Neat | 29 h | 95 |
| 5c | 2e | CH3 (1a) | 6-Cl | H | Neat | 48 h | 94 |
| 6c | 2f | CH3 (1a) | 6-Br | H | Neat | 4 days | 91 |
| 7c | 2g | CH3 (1a) | 5-F | H | Neat | 14 h | 93 |
| 8c | 2h | CH3 (1a) | 5-Cl | H | Neat | 30 h | 94 |
| 9c | 2i | CH3 (1a) | 5-Br | H | Neat | 36 h | 90 |
| 10c | 2j | CH3 (1a) | 5-Me | H | Neat | 60 h | 90 |
| 11c | 2k | CH3 (1a) | 4-Br | H | Neat | 40 h | 88 |
| 12c | 2l | CH3 (1a) | 4,7-Cl2 | H | Neat | 50 h | 92 |
| 13d | 2a | 2-Np (1b) | H | H | THF | 7.5 days | 90 |
| 14d | 2g | 2-Np (1b) | 5-F | H | THF | 6 days | 92 |
| 15d | 2h | 2-Np (1b) | 5-Cl | H | THF | 7 days | 95 |
| 16d | 2h | 1-Np (1c) | 5-Cl | H | THF | 8 days | 85 |
| 17d | 2a | Ph (1d) | H | H | THF | 7 days | 90 |
| 18d | 2g | Ph (1d) | 5-F | H | THF | 6 days | 86 |
| 19d | 2h | Ph (1d) | 5-Cl | H | THF | 6.5 days | 88 |
| 20d | 2g | 4-Cl–Ph (1e) | 5-F | H | THF | 7 days | 85 |
| 21d | 2h | 4-Cl–Ph (1e) | 5-Cl | H | THF | 7 days | 87 |
| 22d | 2h | 4-Me–Ph (1f) | 5-Cl | H | THF | 8 days | 80 |

Here it is important to mention that aromatic ketones were not as reactive as was the case with acetone. When we tried the liquid aromatic ketones (acetophenone) under the above optimized condition the reaction did not reach completion even after 7 days. When the same reaction was conducted in the presence of THF as solvent, we got a relatively higher product yield, at room temperature, in 3 days. Increasing the reaction temperature caused the formation of by-products. However, on increasing the catalyst loading to 50 mg (Table 3, entry 17) from the optimized 25 mg, the reaction was over in 7 days in the case of acetophenone, but for other ketones, it took 6–8 days to give the corresponding aldol adduct in good isolated yield (up to 98%) and excellent selectivity (up to ≥99%). A further increase in the catalyst loading was of no consequence.
Activity of immobilized catalyst in Knoevenagel condensation reactions of isatins
Once the catalytic efficiency of the catalyst IMPC was confirmed with the aldol adducts obtained in high yields, we tried to explore other synthetic methodologies that would offer indole derivatives. As malononitrile has an active methylene moiety, it would be able to give Knoevenagel products. Therefore, we further expanded the scope of our catalyst IMPC to get Knoevenagel products, which are useful intermediates, building blocks as the nitrile group can easily be converted to desired functional groups. Accordingly, the condensation reaction was performed using malononitrile as an active methylene compound with isatin derivatives in the presence of IMPC as a catalyst under reaction conditions optimized for aldol reaction. Amazingly in all the cases, irrespective of the nature of the substituent on isatin, the reaction was completed within a period of 10–25 min to give the corresponding product in 94–97% isolated yields (Table 4).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R2 | R3 | Time [min] | Product | Yieldb [%] |
| a Reaction condition: isatins (0.3 mmol), malononitrile (0.3 mmol), THF (1 mL), catalyst (25 mg), RT.b Isolated yield. | ||||||
| 1 | 2a | H | H | 10 | 4a | 99 |
| 2 | 2b | H | Me | 15 | 4b | 96 |
| 3 | 2c | H | Ph | 10 | 4c | 94 |
| 4 | 2d | 7-Cl | H | 15 | 4d | 98 |
| 5 | 2e | 6-Cl | H | 20 | 4e | 95 |
| 6 | 2f | 6-Br | H | 20 | 4f | 98 |
| 7 | 2g | 5-F | H | 10 | 4g | 97 |
| 8 | 2h | 5-Cl | H | 10 | 4h | 96 |
| 9 | 2i | 5-Br | H | 15 | 4i | 99 |
| 10 | 2j | 5-Me | H | 12 | 4j | 96 |
| 11 | 2k | 5-NO2 | H | 25 | 4k | 94 |

Activity of immobilized catalyst in conjugate addition of acetone to isatylidene malononitriles adducts
We further extended the scope of our present catalyst IMPC in the synthesis of isatylidene malononitriles adducts by the condensation of Knoevenagel products obtained above (as in Table 4) with acetone at room temperature (Table 5). Here again, to begin with we used the catalyst loading of 25 mg per 0.3 mmol of substrate, the conjugate addition reaction took 24 h to complete (Table 5, entry 1). In order to reduce the reaction time we increased the catalyst loading incrementally to 50 mg, 75 mg and 100 mg per 0.3 mmol of the substrate (entries 2–4) and found that 75 mg of catalyst was optimum (entry 4) to give conjugate addition product in 2.5 h reaction in a positive manner.
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Catalyst loading [mg] | Time [h] | Product | Yieldb [%] |
| a Reaction condition: substrate (0.3 mmol), acetone (1 mL), RT.b Isolated yields. | |||||
| 1 | 4a | 25 | 24 | 5a | 86 |
| 2 | 4a | 50 | 18 | 5a | 90 |
| 3 | 4a | 75 | 2.5 | 5a | 95 |
| 4 | 4a | 100 | 2 | 5a | 94 |
| 5 | 4b | 75 | 2.5 | 5b | 90 |
| 6 | 4c | 75 | 2.5 | 5c | 88 |
| 7 | 4d | 75 | 3 | 5d | 92 |
| 8 | 4e | 75 | 7 | 5e | 92 |
| 9 | 4f | 75 | 6 | 5f | 93 |
| 10 | 4g | 75 | 3 | 5g | 92 |
| 11 | 4h | 75 | 3 | 5h | 90 |
| 12 | 4i | 75 | 4 | 5i | 88 |
| 13 | 4j | 75 | 7 | 5j | 85 |
| 14 | 4k | 75 | 5 | 5k | 85 |

As a logical extension of our work we tried to perform Knoevenagal condensation and conjugate addition reaction in one pot using IMPC catalyst by sequentially adding malononitrile and acetone to isatin to get conjugate addition product. However, we observed several side products with low conjugate addition product yield. Therefore, it was better to conduct the two reactions separately with our catalytic system.
Recyclability of IMPC in aldol addition of acetone to isatin
To see the possibility of catalyst IMPC recyclability, we used aldol addition of acetone to isatin under the optimized solvent free condition (Table 2, entry 1). After the reaction was over, the reaction mass was centrifuged and the catalyst was washed sequentially with methylene chloride, acetone and dried at 60 °C under vacuum. The recovered catalyst showed characteristics peaks in IR (see ESI†) as observed in fresh catalyst. The recovered catalyst was used as fresh catalyst in subsequent catalytic runs that showed no sign of deactivation over 10 cycles used (Fig. 7) (for TEM images and Isotherm profile of recycled IMPC, see ESI†).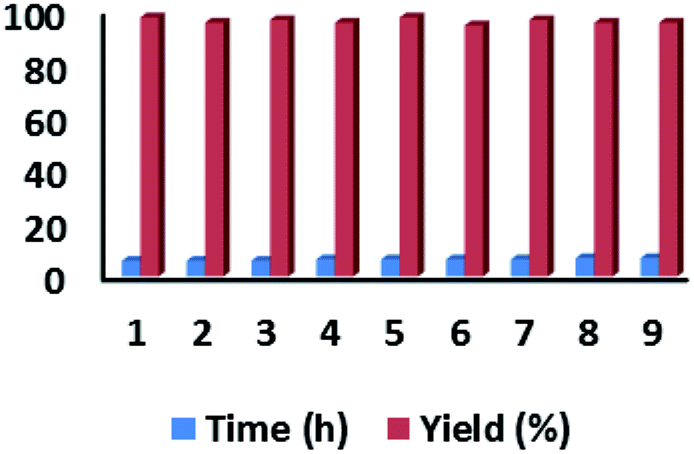 | ||
| Fig. 7 Recycle data of IMPC in aldol addition of acetone to isatin. | ||
Conclusions
We explored the piperazine bound mesoporus silica using triazine (linker), as an efficient catalyst for the aldol, Knoevenagel and conjugate addition of isatidylene derivatives. Though the yield observed in the case of cross aldol reactions was good to excellent, it suffered from long reaction time, whereas the same catalyst showed good results in Knoevenagel and conjugate additions in less reaction time with excellent yields. Currently our group is working on the asymmetric base catalyzed reactions using AFMS as support and the same will be reported in due course of time.Experimental section
Method for the synthesis of AFMS
In a synthetic procedure for the synthesis of highly ordered amino propyl functionalized mesoporous silica (AFMS),24 10 g of triblock, poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (EO20–PO70–EO20) (pluronic P123, mw 5800) was dissolved in 156 g of double distilled water. In the resulting solution, a solution of Na2SiO3·9H2O (27.34 g in 100 g of double distilled water) was added drop-wise. The solution was then stirred at 40 °C until the entire solution became transparent and homogenous. This was followed by the addition of 3-amino propyl triethoxy silane (APTES) (1.73 g). The resulting mixture was further stirred for 30 min, to which conc. HCl (81 g) was added all at once. The mixture after stirring for 1 h was subjected to microwave irradiation under static condition at 100 °C temperature and 300 W operating power for 2 h. The resulting crystalline product was filtered off, washed with warm deionized water (4 × 500 mL) and dried at 60 °C under vacuum. Soxhlet extraction with ethanol over a period of 24 h was done to remove the template. Finally the solid thus obtained was dried at 60 °C under vacuum. The solid was characterized by means of IR-, XRD, diffuse reflectance UV-Vis spectroscopy, SEM, TEM, microanalysis and N2 adsorption–desorption studies.Synthesis of immobilized catalyst IMPC
IMPC was synthesized according to the procedure reported by Shantz et al.21 In a typical procedure, 1 g of AFMS was added to a solution of cyanuric chloride (CC) (1.25 g, 6.78 mmol) and diisopropylethylamine (DIPEA) (8 mmol) in THF (25 mL) under N2 atmosphere. The resulting slurry was stirred for a period of 24 h and filtered. The residue thus obtained was washed sequentially with 50 mL portions of methanol, dichloromethane (DCM) and THF, and transferred to a RBF containing piperazine (0.86 g, 10 mmol) in dry THF (25 mL) under N2 atmosphere. The resulting mass was stirred for 24 h, filtered and washed in the same manner as described above. Finally the functionalized silica material was dried at RT under vacuum for 6 h (Scheme 1) (1.1 g).General method for aldol addition of ketones to isatins
To a 10 mL vial were added catalyst 25 mg, isatin 2a (50 mg, 0.30 mmol) and acetone (1 mL). The resulting mixture was stirred at room temperature until the consumption of isatin, as monitored by TLC, occurred. The catalyst was separated using centrifugation method and washed with acetone (5 × 2 mL). The solution was then evaporated on rotavapor to get the crude aldol product 3aa. The products were then purified by flash chromatography using silica gel. The products thus obtained were confirmed by the NMR with those with published literature.General method for knoevenagal condensation of isatins
To a 10 mL vial were added catalyst 20 mg, isatin 2a (50 mg, 0.30 mmol) and THF 2 mL as solvent. To the resulting mixture, (1 equiv. 20 μL) of malononitrile was added. The red mixture thus obtained was stirred at room temperature until the consumption of isatin, as monitored by TLC, occurred. The catalyst was separated using centrifugation method and washed with THF (5 × 2 mL). The solution was then evaporated on rotavapor to get the crude Knoevenagel products 4a. The obtained crude products were of enough purity (>95%) but further purification was done by flash chromatography using silica gel. The products obtained were confirmed by NMR with those reported in published literature.General method for conjugate addition of acetone to isatylidene malononitriles adducts
To a 10 mL vial were added catalyst 75 mg, malononitrile adduct 4a (58.5 mg, 0.30 mmol) and 1 mL acetone as nucleophile and solvent. The resulting red mixture was stirred, (which changed to light brown) at room temperature until the consumption of malononitrile adduct, as monitored by TLC, occurred. The catalyst was separated using centrifugation method and washed with acetone (5 × 2 mL). The solution was then evaporated on rotavapor to get the crude Isatylidene malononitriles products 5a. The obtained crude products, which solidified on standing, were purified by flash chromatography using silica gel. The products obtained were confirmed by NMR with those reported in published literature.Acknowledgements
The CSIR-CSMCRI registration no. is CSIR-CSMCRI-188/2014. Authors are thankful to CSIR-Indus Magic Project CSC0123 for their financial support. Naveen Gupta is thankful to AcSIR for Ph. D enrolment and DST for INSPIRE fellowship under Doctoral Study. Analytical Discipline and Centralized Instrumental Facility is gratefully acknowledged for providing instrumental facilities.Notes and references
- (a) Y. Q. Tang, I. Sattler, R. Thiericke, S. Grabley and X. Z. Feng, Eur. J. Org. Chem., 2001, 261–267 CrossRef CAS; (b) M. Toyota and M. Ihara, Nat. Prod. Rep., 1998, 15, 327–340 RSC; (c) J. J. Song, J. T. Reeves, F. Gallou, Z. Tan, N. K. Yee and C. H. Senanayake, Chem. Soc. Rev., 2007, 36, 1120–1132 RSC; (d) R. J. Sundberg, The Chemistry of Indoles, Academic, New York, 1970 Search PubMed; (e) S. Lin, Z. Q. Yang, B. H. B. Kwok, M. Koldobskiy, C. M. Crews and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6347–6355 CrossRef CAS PubMed; (f) S. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2002, 41, 512–515 CrossRef CAS; (g) X. Z. Wearing and J. M. Cook, Org. Lett., 2002, 4, 4237–4240 CrossRef CAS PubMed.
- (a) G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1, 1045–1075 RSC; (b) R. Dalpozzo, G. Bartolib and G. Bencivennib, Chem. Soc. Rev., 2012, 41, 7247–7290 RSC; (c) N. Shibata, E. Suzuki, T. Asahi and M. Shiro, J. Am. Chem. Soc., 2001, 123, 7001 CrossRef CAS PubMed; (d) R. Shintani, M. Inoue and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 3353 CrossRef CAS PubMed; (e) Z. Mao and S. W. Baldwin, Org. Lett., 2004, 6, 2425–2428 CrossRef CAS PubMed; (f) Z. Xiao, C. Yu, T. Li, X. S. Wang and C. Yao, Org. Lett., 2014, 16, 3632–3635 CrossRef CAS PubMed; (g) A. Singh, A. L. Loomer and G. P. Roth, Org. Lett., 2012, 14, 5266–5269 CrossRef CAS PubMed.
- (a) W. G. Sumpter and F. M. Miller, Chemistry of Heterocyclic Compounds: Heterocyclic Compounds with Indole and Carbazole Systems, 2008, vol. 8, pp. 196–288 Search PubMed; (b) M. Ishikura, T. Abe, T. Choshib and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC; (c) A. Kumar and S. S. Chimni, RSC Adv., 2012, 2, 9748–9762 RSC , references therein.
- (a) S. Mohammadi, R. Heiran, R. P. Herrera and E. M. López, ChemCatChem, 2013, 5, 2131–2148 CrossRef CAS; (b) N. Lashgari and M. Ziarani, ARKIVOC, 2012, 277–320 CrossRef CAS; (c) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed.
- (a) A. Wurtz, Bull. Soc. Chim. Fr., 1872, 17, 436–442 Search PubMed; (b) A. T. Nielsen and W. J. Houlihan, The Aldol Condensation, Org. React., 2011, 16, 1–438 Search PubMed; (c) B. M. Trost, Comprehensive Organic Synthesis, Pergamon Press, Oxford, 1991, vol. 2, pp. 133–340 Search PubMed; (d) F. Braude and H. G. Lindwall, J. Am. Chem. Soc., 1933, 55, 325–327 CrossRef CAS; (e) S. J. Garden, R. B. da Silva and A. C. Pinto, Tetrahedron, 2002, 58, 8399 CrossRef CAS.
- (a) E. Knoevenagel, Chem. Ber., 1894, 27, 2345 CrossRef; (b) G. Jones, Org. React., 1967, 15, 204 CAS; (c) D. V. Demchuk, M. N. Elinson and G. I. Nikishin, Mendeleev Commun., 2011, 21, 224–225 CrossRef CAS PubMed; (d) L. F. Tietze and U. Beifuss, Comprehensive Organic Synthesis, Pergamon Press, Oxford, 1991, p. 341 Search PubMed; (e) N. Lashgaria, G. M. Ziarania, A. Badiei and P. Gholamzadeh, Eur. J. Chem., 2012, 3, 310–313 CrossRef; (f) P. S. Rao and R. V. Venkataratnam, Tetrahedron Lett., 1991, 32, 582 Search PubMed.
- (a) S. L. Zhu, K. Zhao, X. M. Su and S. J. Ji, Synth. Commun., 2009, 39, 1355 CrossRef CAS; (b) L. Liu, D. Wu, X. Li, S. Wang, H. Li, J. Li and W. Wang, Chem. Commun., 2012, 48, 1692–1694 RSC; (c) H. Zhao, Y. B. Lan, Z. M. Liu, Y. Wang, X. Wang and J. C. Tao, Eur. J. Org. Chem., 2012, 1935–1944 CrossRef CAS.
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed; (b) Y. Zhang and J. S. Panek, Org. Lett., 2009, 11, 3366–3369 CrossRef CAS PubMed; (c) Z. Y. Cao, Y. Zhang, C. B. Ji and J. Zhou, Org. Lett., 2011, 13, 6398–6401 CrossRef CAS PubMed.
- (a) J. F. M. da Silva, S. J. Garden and A. C. Pinto, J. Braz. Chem. Soc., 2001, 12, 273 CrossRef CAS PubMed; (b) F. D. Popp, Adv. Heterocycl. Chem., 1975, 18, 1–58 CrossRef CAS.
- (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726–3748 CrossRef CAS; (b) F. Liu, Chirality, 2013, 25, 675–683 CrossRef CAS PubMed.
- (a) D. C. Hamilton and R. Tooze, Catalyst Separation, Recovery and Recycling, 2006, vol. 30, pp. 9–37 Search PubMed; (b) C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275–3300 CrossRef CAS PubMed; (c) F. Cozzia, Adv. Synth. Catal., 2006, 348, 1367–1390 CrossRef.
- (a) F. X. Liao, Y. G. Wang and Q. Zhu, Synth. Commun., 2014, 44, 161–169 CrossRef CAS; (b) S. Shanmuganathan, L. Greiner and P. D. de María, Tetrahedron Lett., 2010, 51, 6670–6672 CrossRef CAS PubMed; (c) K. Zumbansen, A. Döhring and B. List, Adv. Synth. Catal., 2010, 352, 1135–1138 CrossRef CAS.
- (a) J. Simpson, D. L. Rathbone and D. C. Billington, Tetrahedron Lett., 1999, 40, 7031–7033 CrossRef CAS; (b) G. Panicker, R. Krishnan and K. Sreekumar, Tetrahedron Lett., 2014, 55, 2352–2354 CrossRef PubMed; (c) M. Amirnejad, M. Reza, N. Jamal, H. Tourani and H. Ghafuri, Monatsh. Chem., 2013, 144, 1219–1225 CrossRef CAS; (d) X. Dong, Y. Hui, S. Xie, P. Zhang, G. Zhou and Z. Xie, RSC Adv., 2013, 3, 3222–3226 RSC.
- M. T. Barros and A. M. Faísca Phillips, Eur. J. Org. Chem., 2007, 178–185 CrossRef CAS.
- (a) S. Hanessian and V. Pham, Org. Lett., 2000, 2, 2975–2978 CrossRef CAS PubMed; (b) For heterogenous basic catalyst see; H. Hattori, Chem. Rev., 1995, 95, 537–550 CrossRef CAS.
- (a) C. E. T. Mitchell, S. E. Brenner and S. V. Ley, Chem. Commun., 2005, 5346–5348 RSC; (b) N. Nikbin and P. Watts, Org. Process Res. Dev., 2004, 8, 942 CrossRef CAS; (c) G. Yang, Z. Chen, G. Xu and X. Nie, Catal. Commun., 2004, 5, 75–78 CrossRef CAS PubMed.
- E. Rodrigo, B. G. Alcubilla, R. Sainz, J. L. G. Fierro, R. Ferritto and M. B. Cid, Chem. Commun., 2014, 50, 6270–6273 RSC.
- W. B. Chen, Y. H. Liao, X. L. Du, X. M. Zhang and W. C. Yuan, Green Chem., 2009, 11, 1465–1476 RSC.
- T. Roy, R. I. Kureshy, N. H. Khan, S. H. R. Abdi, A. Sadhukhan and H. C. Bajaj, Tetrahedron, 2012, 68, 6314–6322 CrossRef CAS PubMed.
- J. Roser, K. Kailasan and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef PubMed.
- E. J. Acosta, C. S. Carr, E. E. Simanek and D. F. Shantz, Adv. Mater., 2004, 16, 985–989 CrossRef CAS.
- (a) D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS; (b) M. K. Naskar and M. Eswaramoorthy, J. Chem. Sci., 2008, 120, 181–186 CrossRef CAS PubMed.
- W. C. Chen, S. Y. Wu, H. P. Liu, C. H. Chang, H. Y. Chen, H. Y. Chen, C. H. Ti, Y. C. Chang, F. J. Tsai, K. M. Man, P. L. Liu, F. Y. Lin, J. L. Shen, W. Y. Lin and Y. H. Chen, J. Clin. Lab. Anal., 2010, 24, 92–99 CrossRef CAS PubMed.
- Sujandi, S. E. Park, D. S. Han, S. C. Han, M. J. Jin and T. Ohsuna, Chem. Commun., 2006, 4131–4133 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, solid state NMR of catalyst and characterization data of final products including spectra, TEM images and N2-adsorption–desorption isotherm. See DOI: 10.1039/c5ra00406c |
| This journal is © The Royal Society of Chemistry 2015 |